一種低模板劑用量合成ZSM?22/ZSM?23復合分子篩的方法與流程
本發明涉及一種低模板劑用量合成zsm-22/zsm-23復合分子篩的方法。
背景技術:
隨著環保法規要求的日益嚴格和汽車工業的飛速發展,不斷出現的新型機械設備對高品質潤滑油的需求逐漸增加,質量要求也越來越高。因此,潤滑油質量向低氮、低硫、低灰分,高抗氧化性、高清凈性、分散性,低揮發性、良好黏溫性和低溫流動性的ii、iii類油發展勢在必行,從而使得以溶劑精制和催化脫蠟為主的基礎油生產工藝逐漸被異構脫蠟工藝所替代。異構脫蠟反應是將蠟分子和環烷烴的長側鏈轉化成帶支鏈的烷烴,既降低了產品的傾點,又保留了較高的黏度指數和收率,因此,異構脫蠟技術被稱為近年來石油煉制領域的重大技術進步,其技術核心是加氫異構催化劑,而作為酸性中心的沸石分子篩更是異構催化劑中的重中之重。蠟的主要成分是高熔點的長鏈正構烷烴(碳數最高可達200以上),其傾點高,低溫流動性能差。通過臨氫異構化反應將正構烷烴轉化為支鏈烷烴,可以改善這些性能,得到高品質的潤滑油基礎油。然而,對于蠟中的較長鏈正構烷烴,僅通過臨氫異構過程很難達到理想的傾點,要求部分碳鏈發生斷裂,即要求臨氫異構反應的同時發生適當的裂化反應,才能使潤滑油基礎油的產品性質達到理想指標。因此需要一種具有多級酸性位、孔道結構的分子篩以滿足這種要求。
復合分子篩通常是由兩種或兩種以上分子篩通過化學方法形成的復合晶體。在結構上既具有單一組分的特性,又兼顧自身獨特的結構特征和多級酸性質,在催化反應中表現出不同于純相分子篩的獨有的協同效應和特殊的反應性能。就雙微孔復合分子篩來說,其具有較低的合成成本,較高的水熱穩定性以及可調的酸性能和微結構性能,從而有利于產物選擇性的提高,在石油化工等行業中表現出更潛在的應用價值。
中國發明專利cn1693196、cn1762807、cn1769173及cn1772611公開了zsm-23/zsm-22復合分子篩及制備方法,其方法為在zsm-22分子篩(或zsm-23)的初始凝膠中加入zsm-23分子篩(或zsm-22)種子,得到的混合物進行水熱晶化得到zsm-23/zsm-22復合分子篩。中國發明專利cn103964460公開了一種微孔復合硅鋁分子篩及其制備方法,利用常規溶膠-凝膠模板法水熱晶化合成具有ton/mtt微觀復合拓撲結構的硅鋁分子篩。縱觀此類常規晶化合成方法,合成zsm-22/zsm-23復合分子篩均需要加入大量有機胺作為模板劑,而這些模板劑最終均需要通過焙燒分解成氣體排放,這不僅提高了沸石分子篩的合成成本,同時在焙燒過程中也會相應產生大量的有害廢氣,直接影響環境安全。
技術實現要素:
本發明的目的是提供一種低成本,低污染的合成zsm-22/zsm-23復合分子篩的方法。
本發明提供的低模板劑用量合成zsm-22/zsm-23復合分子篩的方法,包括如下步驟:
(1)將無機堿源加入去離子水中,攪拌完全溶解后加入模板劑a、鋁源或硼源,繼續攪拌至混合物至完全溶解后,再加入模板劑b及硅源,形成具有如下摩爾配比的第一份初始凝膠混合物,硅源以sio2計,鋁源以al2o3計,硼源以b2o3計,堿源以oh-計,sio2:al2o3或b2o3:模板劑a:模板劑b:oh-:去離子水=1:0.2-0.001:0.05-5.0:0.05-5.0:0.005-2.0:10.0-200.0;
(2)將步驟(1)所得的第一份初始凝膠混合物放置于超聲波裝置,在25-100℃,超聲波頻率40-60khz,超聲波功率100-2500w條件下進行超聲振蕩10-180分鐘,得到一次超聲混合物;
(3)將無機堿源加入去離子水中,攪拌完全溶解后加入鋁源或硼源,繼續攪拌至混合物至完全溶解后,再加入硅源,形成具有如下摩爾配比的第二份初始凝膠混合物,硅源以sio2計,鋁源以al2o3計,硼源以b2o3計,堿源以oh-計,sio2:al2o3或b2o3:oh-:去離子水=1:0.2-0.001:0.005-2.0:10.0-200.0;
(4)以sio2計,按第一份初始凝膠混合物與第二份初始凝膠摩爾比例為5-50:95-50,將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25-100℃,超聲波頻率40-60khz,超聲波功率100-2500w條件下進行超聲振蕩10-180分鐘,得到的二次超聲混合物轉入晶化釜中,在140-200℃、晶化12-240h;
(5)待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于400-600℃焙燒1-72h,即得到zsm-22/zsm-23復合分子篩。
如上所述的堿源是氫氧化鈉、氫氧化鉀中的一種或兩種的組合。
如上所述的模板劑a是乙甲胺、二乙胺、1,6-己二胺、n-異丙基二乙烯三胺、1-甲基丁胺中、乙二胺的一種或兩種的混合。
如上所述的鋁源是擬薄水鋁石、氯化鋁、硫酸鋁、硝酸鋁、異丙醇鋁、鋁酸鈉中一種或任意幾種的組合。
如上所述的硼源是硼酸鈉、硼酸鉀、硼酸中一種或任意幾種的組合。
如上所述的模板劑b是二甲胺、二甲基甲酰胺、正丙胺、異丙胺、正丁胺、異丁胺、新戊胺、三甲胺、n-異丙基-1,3-丙二胺和n,n,n',n',-四甲基二丙烯三胺等中的一種或兩種的混合。
如上所述的硅源是硅溶膠、硅凝膠、正硅酸甲酯、正硅酸乙酯、硅酸鈉、白炭黑中的一種或任意幾種的組合。
如上所述的步驟(4)中晶化方式可以是自身壓力下靜態晶化,也可以是動態晶化。
本發明所制得的zsm-22/zsm-23復合分子篩中zsm-22:zsm-23摩爾比為10-90:90-10,具有結晶度高,sio2:al2o3或b2o3摩爾比例可調范圍廣(可在5-1000內調整),bet比表面大于250m2/g,顆粒為長度<2μm,外徑20-50nm均勻、細長棒狀物,無雜晶相等特征。
本發明提供的方法與現有方法相比,具有如下優點:
(1)與現有合成方法相比,采用本發明模板劑用量可減少至原來的5-50wt.%,極大地降低分子篩合成成本及對環境的影響。
(2)采用本發明提供的合成方法,一次超聲振蕩可快速誘導初始凝膠凝聚形成晶核,并避免晶核團聚長大,二次超聲振蕩可加快誘導凝膠溶液在晶核表面凝聚成晶,可有效避免雜晶的出現,因此與現有合成方法相比,所獲得的復合分子篩無雜晶相,粒度小而均勻,制備重現性好。
(3)采用本發明提供的方法能夠在更寬范圍內調變硅鋁比,制得的分子篩具有多級可調變的酸性位、多級微型孔道結構的特性,特別適合含有長鏈烴的蠟對分子篩的要求,對于長鏈烴的臨氫異構降凝作用具有優異的催化性能,適應性極高,在加氫裂化和異構化等方面具有極大的潛在應用價值。
附圖說明
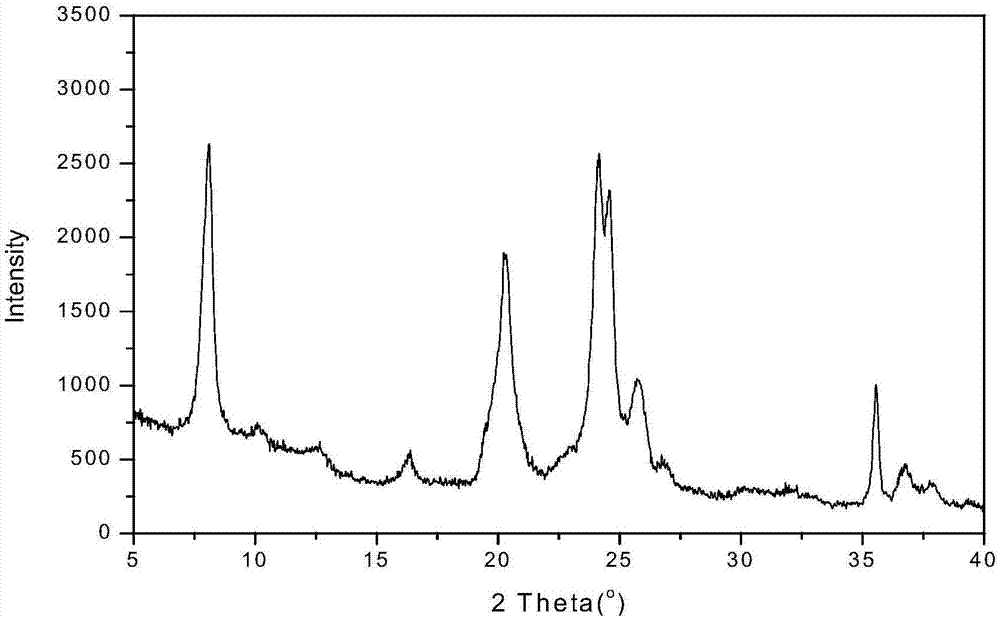
圖1是實施例2合成zsm-22/zsm-23復合分子篩的xrd譜圖。
圖2是實施例6合成zsm-22/zsm-23復合分子篩的xrd譜圖。
具體實施方式
下面通過實施例詳述本發明,但本發明并不局限于這些實施例。
實施例1:
將氫氧化鉀加入去離子水中,攪拌完全溶解后先后加入乙甲胺、鋁酸鈉,繼續攪拌至混合物完全溶解后,按順序加入二甲胺及硅溶膠,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:乙甲胺:二甲胺:oh-:去離子水=1:0.2:0.05:5.0:0.005:10.0;向所得的初始凝膠混合物放置于超聲波裝置,在25℃,超聲波頻率50khz,超聲波功率100w條件下進行超聲振蕩180分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為5:95(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率50khz,超聲波功率100w條件下進行超聲振蕩180分鐘,得到的二次超聲混合物轉入晶化釜中,然后在140℃、自生壓力下晶化240h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于400℃焙燒72h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=6,bet比表面275.5m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=10:90,sem結果分子篩形貌為長度1.5~2μm,外徑20~30nm的細長棒狀物。
實施例2:
將氫氧化鉀加入去離子水中,攪拌完全溶解后先后加入二乙胺、擬薄水鋁石,繼續攪拌至混合物完全溶解后,按順序加入二甲基甲酰胺、正丙胺及硅凝膠,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:二乙胺:二甲基甲酰胺+正丙胺:oh-:去離子水=1:0.1:0.1:4.0(二甲基甲酰胺:正丙胺=1:3,摩爾比):0.01:30.0;向所得的初始凝膠混合物放置于超聲波裝置,在30℃,超聲波頻率40khz,超聲波功率500w條件下進行超聲振蕩120分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為10:90(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在30℃,超聲波頻率40khz,超聲波功率500w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在150℃、自生壓力下晶化120h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于450℃焙燒36h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=9,bet比表面267.3m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=30:70,sem結果分子篩形貌為長度1.5~2μm,外徑25~30nm的細長棒狀物。
實施例3:
將氫氧化鉀加入去離子水中,攪拌完全溶解后先后加入1,6-己二胺、氯化鋁,繼續攪拌至混合物完全溶解后,按順序加入異丙胺、正硅酸甲酯及正硅酸乙酯,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:1,6-己二胺:異丙胺:oh-:去離子水=1(正硅酸甲酯:正硅酸乙酯=1:1,摩爾比,以sio2計):0.05:0.2:3.0:0.02:20.0;向所得的初始凝膠混合物放置于超聲波裝置,在40℃,超聲波頻率60khz,超聲波功率1000w條件下進行超聲振蕩60分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為20:80(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在40℃,超聲波頻率60khz,超聲波功率1000w條件下進行超聲振蕩60分鐘,得到的二次超聲混合物轉入晶化釜中,然后在150℃、自生壓力下晶化64h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于500℃焙燒24h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=18,bet比表面282.9m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=25:75,sem結果分子篩形貌為長度1~2μm,外徑20~30nm的細長棒狀物。
實施例4:
將氫氧化鉀及氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入n-異丙基二乙烯三胺、硫酸鋁及硝酸鋁,繼續攪拌至混合物完全溶解后,按順序加入正丁胺、異丁胺及正硅酸乙酯,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:n-異丙基二乙烯三胺:正丁胺+異丁胺:oh-:去離子水=1:0.02(硫酸鋁:硝酸鋁=1:1,摩爾比,以al2o3計):0.5:2.5(正丁胺:異丁胺=1:1.5,摩爾比):0.05(氫氧化鉀:氫氧化鈉=1:1,摩爾比):50.0;向所得的初始凝膠混合物放置于超聲波裝置,在50℃,超聲波頻率60khz,超聲波功率2000w條件下進行超聲振蕩30分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為30:70(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在50℃,超聲波頻率60khz,超聲波功率2000w條件下進行超聲振蕩30分鐘,得到的二次超聲混合物轉入晶化釜中,然后在160℃、自生壓力下晶化48h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于500℃焙燒12h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=47,bet比表面254.7m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=40:60,sem結果分子篩形貌為長度1.5~2μm,外徑35~40nm的細長棒狀物。
實施例5:
將氫氧化鉀及氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入1-甲基丁胺、硝酸鋁,繼續攪拌至混合物完全溶解后,按順序加入特戊胺及白炭黑,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:1-甲基丁胺:特戊胺:oh-:去離子水=1:0.01:1.0:1.0:0.1(氫氧化鉀:氫氧化鈉=1:3,摩爾比):70.0;向所得的初始凝膠混合物放置于超聲波裝置,在70℃,超聲波頻率50khz,超聲波功率2500w條件下進行超聲振蕩10分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為40:60(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在70℃,超聲波頻率50khz,超聲波功率2500w條件下進行超聲振蕩10分鐘,得到的二次超聲混合物轉入晶化釜中,然后在170℃、自生壓力下晶化36h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于550℃焙燒6h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=79,bet比表面263.6m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=55:45,sem結果分子篩形貌為長度1.5~2μm,外徑35~45nm的細長棒狀物。
實施例6:
將氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入乙二胺、異丙醇鋁,繼續攪拌至混合物完全溶解后,按順序加入三甲胺及白炭黑,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:乙二胺:三甲胺:oh-:去離子水=1:0.005:2.0:0.5:0.2:100.0;向所得的初始凝膠混合物放置于超聲波裝置,在100℃,超聲波頻率50khz,超聲波功率500w條件下進行超聲振蕩30分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為50:50(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在100℃,超聲波頻率50khz,超聲波功率500w條件下進行超聲振蕩30分鐘,得到的二次超聲混合物轉入晶化釜中,然后在180℃、自生壓力下晶化24h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于550℃焙燒6h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=179,bet比表面273.2m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=75:25,sem結果分子篩形貌為長度1.5~2μm,外徑35~45nm的細長棒狀物。
實施例7:
將氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入乙二胺、n-異丙基二乙烯三胺、擬薄水鋁石、硫酸鋁,繼續攪拌至混合物完全溶解后,按順序加入n-異丙基-1,3-丙二胺及硅酸鈉,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:乙二胺+n-異丙基二乙烯三胺:n-異丙基-1,3-丙二胺:oh-:去離子水=1:0.002(擬薄水鋁石:硫酸鋁=1:1,摩爾比,以al2o3計):5.0(乙二胺:n-異丙基二乙烯三胺=4:1,摩爾比):0.1:0.5:100.0;向所得的初始凝膠混合物放置于超聲波裝置,在25℃,超聲波頻率40khz,超聲波功率1000w條件下進行超聲振蕩60分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為25:75(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在100℃,超聲波頻率50khz,超聲波功率500w條件下進行超聲振蕩30分鐘,得到的二次超聲混合物轉入晶化釜中,然后在200℃、自生壓力下晶化12h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于600℃焙燒2h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=456,bet比表面271.1m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=65:35,sem結果分子篩形貌為長度1.5~2μm,外徑30~45nm的細長棒狀物。
實施例8:
將氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入二乙胺、1-甲基丁胺、鋁酸鈉,繼續攪拌至混合物完全溶解后,按順序加入n,n,n',n',-四甲基二丙烯三胺、硅酸鈉及硅溶膠,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:二乙胺+1-甲基丁胺:n,n,n',n',-四甲基二丙烯三胺:oh-:去離子水=1(硅酸鈉:硅溶膠=1:1,摩爾比,以sio2計):0.001:1.0(二乙胺:1-甲基丁胺=3:1,摩爾比):0.05:1.0:200.0;向所得的初始凝膠混合物放置于超聲波裝置,在50℃,超聲波頻率40khz,超聲波功率1500w條件下進行超聲振蕩45分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為10:90(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率60khz,超聲波功率100w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在170℃、自生壓力下晶化64h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于600℃焙燒1h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=1000,bet比表面266.4m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=90:10,sem結果分子篩形貌為長度1.5~2μm,外徑30~45nm的細長棒狀物。
實施例9:
將氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入乙二胺、硫酸鋁,繼續攪拌至混合物完全溶解后,按順序加入乙胺及白炭黑,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,鋁源以al2o3計,堿源以oh-計),sio2:al2o3:乙二胺:乙胺:oh-:去離子水=1:0.005:1.0:2.0:2.0:75.0;向所得的初始凝膠混合物放置于超聲波裝置,在60℃,超聲波頻率50khz,超聲波功率500w條件下進行超聲振蕩60分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為5:95(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率60khz,超聲波功率100w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在160℃、自生壓力下晶化64h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于500℃焙燒3h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/al2o3摩爾比=176,bet比表面264.5m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=50:50,sem結果分子篩形貌為長度1.5~2μm,外徑30~45nm的細長棒狀物。
實施例10:
將氫氧化鉀加入去離子水中,攪拌完全溶解后先后加入乙甲胺、硼酸鈉,繼續攪拌至混合物完全溶解后,按順序加入二甲胺及硅溶膠,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,硼源以b2o3計,堿源以oh-計),sio2:b2o3:乙甲胺:二甲胺:oh-:去離子水=1:0.2:0.05:5.0:0.005:10.0;向所得的初始凝膠混合物放置于超聲波裝置,在60℃,超聲波頻率50khz,超聲波功率500w條件下進行超聲振蕩60分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為5:95(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率60khz,超聲波功率100w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在170℃、自生壓力下晶化48h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于400℃焙燒72h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/b2o3摩爾比=5,bet比表面252.8m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=10:90,sem結果分子篩形貌為長度1.5~2μm,外徑40~50nm的細長棒狀物。
實施例11:
將氫氧化鉀加入去離子水中,攪拌完全溶解后先后加入二乙胺、硼酸鉀,繼續攪拌至混合物完全溶解后,按順序加入二甲基甲酰胺、正丙胺及硅凝膠,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,硼源以b2o3計,堿源以oh-計),sio2:b2o3:二乙胺:二甲基甲酰胺+正丙胺:oh-:去離子水=1:0.1:0.1:4.0(二甲基甲酰胺:正丙胺=1:3,摩爾比):0.01:30.0;向所得的初始凝膠混合物放置于超聲波裝置,在60℃,超聲波頻率50khz,超聲波功率500w條件下進行超聲振蕩60分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為5:95(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率60khz,超聲波功率100w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在150℃、自生壓力下晶化120h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于450℃焙燒36h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/b2o3摩爾比=8,bet比表面276.0m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=10:90,sem結果分子篩形貌為長度1~1.5μm,外徑20~30nm的細長棒狀物。
實施例12:
將氫氧化鉀加入去離子水中,攪拌完全溶解后先后加入1,6-己二胺、硼酸,繼續攪拌至混合物完全溶解后,按順序加入異丙胺、正硅酸甲酯及正硅酸乙酯,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,硼源以b2o3計,堿源以oh-計),sio2:b2o3:1,6-己二胺:異丙胺:oh-:去離子水=1(正硅酸甲酯:正硅酸乙酯=1:1,摩爾比,以sio2計):0.05:0.2:3.0:0.02:20.0;向所得的初始凝膠混合物放置于超聲波裝置,在30℃,超聲波頻率50khz,超聲波功率1000w條件下進行超聲振蕩30分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為10:90(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率60khz,超聲波功率100w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在150℃、自生壓力下晶化64h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于500℃焙燒24h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/b2o3摩爾比=18.2,bet比表面261.5m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=15:85,sem結果分子篩形貌為長度1~1.5μm,外徑20~40nm的細長棒狀物。
實施例13:
將氫氧化鉀及氫氧化鈉加入去離子水中,攪拌完全溶解后先后加入n-異丙基二乙烯三胺、硼酸鈉及硼酸,繼續攪拌至混合物完全溶解后,按順序加入正丁胺、異丁胺及正硅酸乙酯,形成具有如下摩爾配比的初始凝膠混合物(硅源以sio2計,硼源以b2o3計,堿源以oh-計),sio2:b2o3:n-異丙基二乙烯三胺:正丁胺+異丁胺:oh-:去離子水=1:0.02(硼酸鈉:硼酸=1:1,摩爾比,以b2o3計):0.5:2.5(正丁胺:異丁胺=1:4,摩爾比):0.05(氫氧化鉀:氫氧化鈉=2:1,摩爾比):50.0;向所得的初始凝膠混合物放置于超聲波裝置,在30℃,超聲波頻率50khz,超聲波功率1000w條件下進行超聲振蕩30分鐘,得到一次超聲混合物;按上述的原料配比重新配制第二份初始凝膠混合物(不添加模板劑),按第一份初始凝膠與第二份初始凝膠摩爾比例為10:90(以sio2計);將一次超聲混合物加入到第二份初始凝膠混合物中,攪拌均勻后置于超聲波裝置,在25℃,超聲波頻率60khz,超聲波功率100w條件下進行超聲振蕩120分鐘,得到的二次超聲混合物轉入晶化釜中,然后在160℃、自生壓力下晶化48h;待晶化完成后,將固體產物過濾分離,用去離子水洗滌至中性,干燥后于500℃焙燒12h,即得到zsm-22/zsm-23復合分子篩。icp分析sio2/b2o3摩爾比=46.5,bet比表面262.8m2/g,xrd表征復合分子篩中zsm-22:zsm-23摩爾比=15:85,sem結果分子篩形貌為長度1~1.5μm,外徑20~40nm的細長棒狀物。
實施例14:
將如上實施例所制得的復合分子篩經離子交換轉為銨型,干燥焙燒后浸漬貴金屬pt,負載量為分子篩的0.5wt.%,經干燥焙燒后得到復合分子篩負載貴金屬pt的加氫異構催化劑,在臨氫條件下以正十六烷為模型反應物進行加氫反應,反應溫度350℃,壓力4.0mpa,液體空速1.5h-1,h2/c16=700(體積比),結果如下表所示:
如上表所示,以本方法制得的復合分子篩為酸性載體制備加氫異構催化劑,表現出優異的異構化性能,在優選的條件下,i-c16收率可達到81.6%。
- 還沒有人留言評論。精彩留言會獲得點贊!