一種金屬摻雜氧化鋅液相均勻分散體的制備方法與流程
本發明屬于金屬材料制備技術領域。
背景技術:
有機太陽能電池由于其在產品制造中具有的輕薄、柔性、易于在低溫和非真空環境中大面積制備等特點,具有廣闊的商業應用前景。界面層具有調節能級,提高電荷的傳輸和收集,提高活性層對光的吸收和提高器件穩定性的功能。所以,為滿足社會發展對太陽能電池的高性能要求,對界面層的研究勢在必行。氧化鋅(zno)是研究最廣泛的并且具有突出應用價值的界面層材料,因具有匹配的能級、高的電子遷移率、好的透光性和環境穩定性以及低成本的優點,同時,通過修飾摻雜的高電導率的zno還能低溫溶液加工制備,并適用于卷對卷印刷制備柔性襯底的電池,在聚合物電池中得到廣泛研究應用。然而,由于zno表面存在不同程度的缺陷,這些缺陷成為電子捕獲中心,極大地抑制了光生載流子的收集,影響了它的進一步應用。
改善低溫溶液法制備zno導致其表面缺陷多且導電率低的研究方法中,目前還是以元素摻雜為主,采用主族元素來對zno進行摻雜已見大量文獻報道,如第ⅱ主族的鎂(mg)、鍶(sr)和鋇(ba),以及第ⅲ主族元素的鎵(ga)、銦(in)和鋁(al)等,這些元素摻雜是作為n-型摻雜來替代zn的位置和收集自由電子。thambidurai等(j.mater.chem.c,2013,1,8161-8166)采用溶膠-凝膠旋涂的方法制備ga摻雜zno,即gzo納米晶薄膜,以該納米晶薄膜為電子傳輸層基于ptb7:pc71bm的聚合物太陽電池器件效率為7.34%。wang等(j.mater.chem.c,2016,4,10820-10826)制備了高導電性、高穩定性的gzo納米粒子用于ptb7-th:pc71bm體系中,效率達到9.83%,且當其厚度從15nm增長到96nm時,器件性能并沒有發生明顯衰減。jagadamma等(adv.energymater.,2015,1500204)采用氨水處理azo納米晶,在125℃較低溫度下溶液加工制備azo陰極緩沖層,基于ptb7:pc71bm活性層,在玻璃基底上得到10%的效率,在柔性基底上得到8%的效率,并且當azo的厚度從22nm增加至75nm時,器件效率僅從8.8%降至8.1%。liu等(adv.mater.,2016,28,7405-7412)通過低溫溶液發制備了azo前驅體溶液,在反應過程中向體系中加入乙醇胺作為配體,將這種azo作為陰極界面層應用于ptb7-th:pc71bm體系中,器件效率達到10.42%(此時azo厚度約為10nm),并且當膜厚從10nm增長到120nm時,器件效率能夠接近9%。但是,這種在azo納米粒子生長的同時引入與鋅鹽摩爾比為1:1的乙醇胺配體來提高分散性的方法,存在配體引入量過多且難以去除的弊端,從而影響azo的導電性。中國專利cn105621475a公布的一種金屬摻雜氧化鋅液相透明分散體及制備方法中也采用了加入表面活性劑改性azo的粒徑和分散性,并且加入量為理論產物中azo質量的1~10%。眾所周知,制備具有較高的電導率的納米粒子分散體應用于厚度不敏感的界面層,對實現柔性可拉伸太陽能電池器件的制備以及大面積卷對卷印刷有著重要的推動作用。目前國內研究者在探討印刷技術應用于聚合物太陽能電池方面鮮有報道,大面積利用低溫溶液法制備分散穩定且具有高電導率和透光性的導電墨水技術領域的研究還比較少。
因此,需要通過溶液法制備分散穩定且具有高電導率和透光性的金屬摻雜氧化鋅導電墨水。本發明涉及一種高導電性、高穩定性的金屬摻雜氧化鋅液相均勻分散體的制備方法。該方法主要是先通過溶液法在60~80℃下低溫合成金屬摻雜氧化鋅納米粒子,然后將其分散在醇溶劑中,最后加入微量含氨基的表面活性劑來提高金屬摻雜氧化鋅分散體系的穩定性。而且通過實驗證明,由于金屬摻雜以及氨基與納米粒子之間形成氫鍵作用,使得金屬摻雜氧化鋅分散體的導電性和分散穩定性得到有效地提高,可以應用于太陽能電池緩沖層的溶液印刷加工。
技術實現要素:
本發明的目的是提供一種金屬摻雜氧化鋅液相均勻分散體的制備方法,在醇相體系中,采用鋅鹽作為反應源,金屬鹽作為摻雜源,采用低溫、溶液法制備了均勻分散且高導電性的金屬摻雜納米粒子的方法。
本發明是通過以下技術方案實現的。
本發明的所述的一種金屬摻雜氧化鋅液相均勻分散體的制備方法,包括以下步驟:
a)將鋅鹽與金屬鹽加入氮氣瓶中,再加入甲醇制得混合鹽溶液;
b)將堿溶于甲醇溶液中,超聲將其溶解,制得堿液;
c)在氮氣保護下,將步驟a)中所得溶液加熱到60℃,劇烈攪拌;
d)將步驟b)中所得溶液裝入恒壓滴液漏斗中,60℃下,將其逐滴滴入步驟a)中所得溶液中,約20~45min滴完;
e)將步驟d)中所得混合溶液在80℃反應2.5~3.0h,得到淡藍色半透明溶液,即為所制備的金屬摻雜氧化鋅納米粒子溶液;
f)將步驟e)得到的淡藍色溶液進行離心,上層清液丟棄,待步驟e)中所得溶液離心完畢后,取離心管底部物質,向其中加入一定量的甲醇,超聲,使得其完全分散在甲醇中,離心,取底部膠狀物質。
g)將步驟f)所得膠狀物質用有機溶劑進行分散,最后分別加入一定量的含不同氨基數目的表面活性劑,所得淡藍色透明狀液體為高導電性和高穩定性的金屬摻雜氧化鋅液相均勻分散體。
本發明步驟a)所述鋅鹽為醋酸鋅、硫酸鋅、硝酸鋅或乙酰丙酮鋅。所述金屬鹽為硝酸鋁,異丙醇鋁,醋酸鋁,硝酸鎵或硝酸銦;所述鋅鹽與金屬鹽的摩爾比為1:0.02。
本發明步驟a)所述的鋅鹽與步驟b)中堿的質量比為1:1.5~1:1.8。
本發明步驟f)中所述的離心速度為8000轉/min,離心時間為10min。
本發明步驟g)所述的有機溶劑選自下列物質中的一種或多種:甲醇、乙醇、異丙醇或正丁醇;所述的含不同氨基數目的表面活性劑包括:乙醇胺、乙二胺、二乙烯三胺、三乙烯四胺、聚乙烯亞胺、油酸銨、檸檬酸胺或氨水,表面活性劑的加入量為表面活性劑占實際產物中金屬摻雜氧化鋅納米粒子質量的4.5×10-3%~5.4×10-3%。
本發明得到的金屬摻雜氧化鋅液相均勻分散體的濃度為10~20mg/ml。
本發明的方法簡單、易行、可控。本發明主要是該方法主要是先通過溶液法在80℃下低溫合成金屬摻雜氧化鋅納米粒子,然后再通過表面活性劑的加入以獲得高穩定性、高導電性的納米材料。表面活性劑中的氨基和羥基能夠與納米粒子表面的氧形成氫鍵,由于氫鍵的作用,使得納米粒子之間團聚減少,得到高穩定性的納米粒子溶液。另外,氨基中的富電子可以實現向納米粒子的轉移,這有利于進一步提高納米粒子的導電性,增加了納米材料的電子遷移率,作為太陽能電池的陰極界面層時,表面活性劑中的功能性基團還可調節界面層與受體pcbm之間形成更加匹配的能級,使得電荷傳輸和收集效率得到提高。本發明所得到的納米材料具有較高的穩定性的同時又具有高的導電性,這有利于制備厚度不敏感的界面層,對于實現制備柔性可拉伸器件和卷對卷大面積印刷具有重要意義。
本發明的有益效果如下:本發明的金屬摻雜氧化鋅透明分散體的制備溫度不超過80℃,制備過程簡單,節能環保。本發明將金屬摻雜氧化鋅納米粒子分散在醇相中,然后利用表面活性劑對其進行改性,制得的納米粒子分散體濃度達到20mg/ml,可見光透過率達90%,顆粒粒徑分散均勻,顆粒直徑為10~20nm,結晶度好,產品穩定性好,靜置超過3個月無沉降。
另外,由于所制備的溶液濃度較大,在制備薄膜時,可以大大減少涂膜次數,每次涂膜之間無需高溫鍛燒過程,在簡化實驗流程的同時也起到節能環保的作用。
附圖說明
圖1中(a)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子(azo)分散液與實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子分散液(azo-ea)、實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子分散液(azo-eda)、實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子分散液(azo-deta)、實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子分散液(azo-teta)放置24h的光學照片。
(b)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子(azo)分散液與實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子分散液(azo-ea)、實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子分散液(azo-eda)、實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子分散液(azo-deta)、實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子分散液(azo-teta)放置1個月的光學照片。
(c)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子(azo)分散液與實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子分散液(azo-ea)、實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子分散液(azo-eda)、實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子分散液(azo-deta)、實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子分散液(azo-teta)放置2個月的光學照片。
(d)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子(azo)分散液與實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子分散液(azo-ea)、實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子分散液(azo-eda)、實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子分散液(azo-deta)、實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子分散液(azo-teta)放置3個月的光學照片。
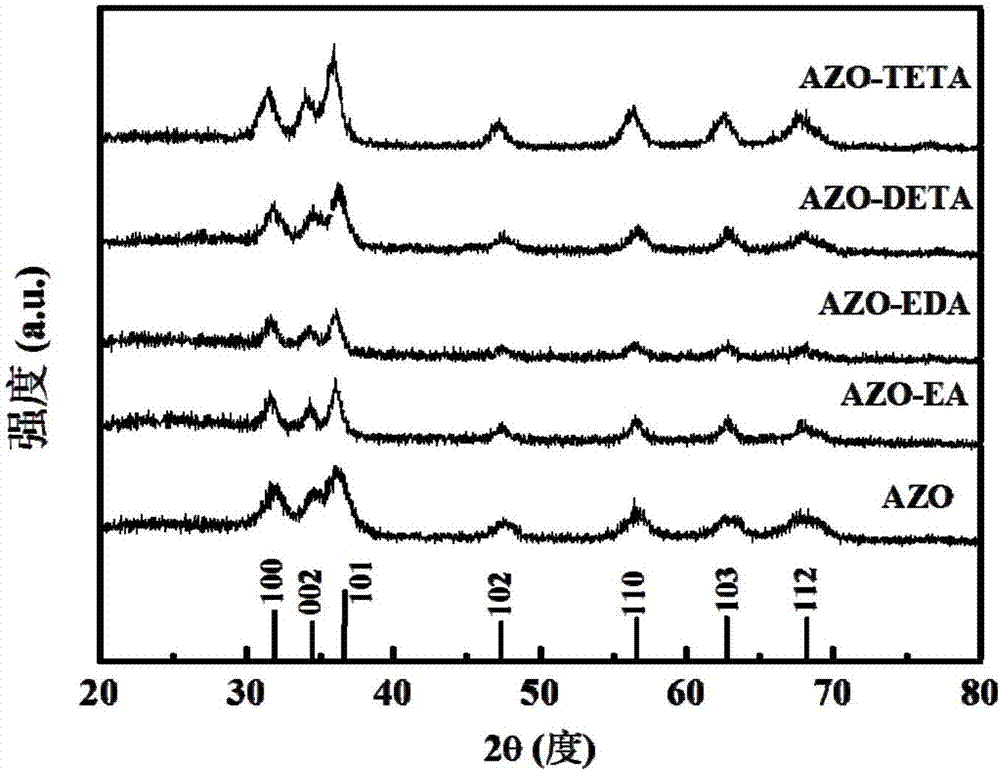
圖2為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子粉末(azo)與實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子粉末(azo-ea)、實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子粉末(azo-eda)、實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子粉末(azo-deta)、實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子粉末(azo-teta)的x射線衍射(xrd)圖譜。
圖3中(a)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子(azo)薄膜的掃描電子顯微鏡(sem)照片。(b)為實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子(azo-ea)薄膜的掃描電子顯微鏡(sem)照片。(c)為實施例3制備的加入表面活性劑乙醇胺(eda)的金屬摻雜氧化鋅納米粒子(azo-eda)薄膜的掃描電子顯微鏡(sem)照片。(d)為實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子(azo-deta)薄膜的掃描電子顯微鏡(sem)照片。(e)為實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子(azo-teta)薄膜的掃描電子顯微鏡(sem)照片。
圖4中(a)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子薄膜(azo)的導電原子力顯微鏡(c-afm)照片。(b)為本發明實施例1制備的未加入表面活性劑的金屬摻雜氧化鋅納米粒子薄膜(azo)的導電原子力顯微鏡(c-afm)的電流分布圖。
圖5中(a)為本發明實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子薄膜(azo-ea)的導電原子力顯微鏡(c-afm)照片。(b)為本發明實施例2制備的加入表面活性劑乙醇胺(ea)的金屬摻雜氧化鋅納米粒子薄膜(azo-ea)的導電原子力顯微鏡(c-afm)的電流分布圖。
圖6中(a)為本發明實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子薄膜(azo-eda)的導電原子力顯微鏡(c-afm)照片。(b)為本發明實施例3制備的加入表面活性劑乙二胺(eda)的金屬摻雜氧化鋅納米粒子薄膜(azo-eda)的導電原子力顯微鏡(c-afm)的電流分布圖。
圖7中(a)為本發明實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子薄膜(azo-deta)的導電原子力顯微鏡(c-afm)照片。(b)為本發明實施例4制備的加入表面活性劑二乙烯三胺(deta)的金屬摻雜氧化鋅納米粒子薄膜(azo-deta)的導電原子力顯微鏡(c-afm)的電流分布圖。
圖8中(a)為本發明實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子薄膜(azo-teta)的導電原子力顯微鏡(c-afm)照片。(b)為本發明實施例5制備的加入表面活性劑三乙烯四胺(teta)的金屬摻雜氧化鋅納米粒子薄膜(azo-teta)的導電原子力顯微鏡(c-afm)的電流分布圖。
具體實施方式
本發明將通過以下實施例作進一步說明。
實施例1。
a)稱取6.15g二水合醋酸鋅[zn(ch3coo)2·2h2o]、0.2g九水合硝酸鋁[al(no3)3·9h2o]加入氮氣瓶中,并加入275ml甲醇將其溶解;
b)稱取2.4g氫氧化鉀(koh)加入125ml甲醇中,超聲將其溶解;
c)在氮氣保護下,將步驟a)中所得溶液加熱到60℃,劇烈攪拌;
d)將步驟b)中所得溶液裝入恒壓滴液漏斗中,60℃下,將koh(aq)逐滴滴入步驟a)中所得溶液中,約45min滴完;
e)將步驟d)中所得混合溶液在80℃回流反應3.0h,得到淡藍色半透明溶液,即為所制備的鋁摻雜氧化鋅納米粒子溶液;
f)將步驟e)得到的淡藍色溶液進行離心,取離心管底部膠狀物質,待步驟e)中所得溶液離心完畢后,加入一定量的甲醇,超聲,使得納米粒子完全分散在甲醇中,離心,取底部膠狀物質。
g)將步驟f)所得膠狀物質用有機溶液分散,最終所得納米粒子分散液10ml,濃度為20mg/ml,最終所得產物淡藍色透明狀液體為本發明的azo納米粒子分散液。
實施例2。
a)稱取6.15g二水合醋酸鋅[zn(ch3coo)2·2h2o]、0.2g九水合硝酸鋁[al(no3)3·9h2o]加入氮氣瓶中,并加入275ml甲醇將其溶解;
b)稱取2.4g氫氧化鉀(koh)加入125ml甲醇中,超聲將其溶解;
c)在氮氣保護下,將步驟a)中所得溶液加熱到60℃,劇烈攪拌;
d)將步驟b)中所得溶液裝入恒壓滴液漏斗中,60℃下,將koh(aq)逐滴滴入步驟a)中所得溶液中,約45min滴完;
e)將步驟d)中所得混合溶液在80℃回流反應3.0h,得到淡藍色半透明溶液,即為所制備的鋁摻雜氧化鋅納米粒子溶液;
f)將步驟e)得到的淡藍色溶液進行離心,取離心管底部膠狀物質,待步驟e)中所得溶液離心完畢后,加入一定量的甲醇,超聲,使得納米粒子完全分散在甲醇中,離心,取底部膠狀物質。
g)將步驟f)所得膠狀物質用有機溶液分散,最終所得納米粒子分散液10ml,濃度為20mg/ml,最后向其中加入乙醇胺(ea),向其中加入10μl乙醇胺,最終所得產物淡藍色透明狀液體為本發明的azo納米粒子分散液。
實施例3。
a)稱取1.23g二水合醋酸鋅[zn(ch3coo)2·2h2o]、0.04g九水合硝酸鋁[al(no3)3·9h2o]加入氮氣瓶中,并加入55ml甲醇將其溶解;
b)稱取0.48g氫氧化鉀(koh)加入25ml甲醇中,超聲將其溶解;
c)在氮氣保護下,將步驟a)中所得溶液加熱到60℃,劇烈攪拌;
d)將步驟b)中所得溶液裝入恒壓滴液漏斗中,60℃下,將koh(aq)滴滴入步驟a)中所得溶液中,約20min滴完;
e)將步驟d)中所得混合溶液在80℃回流反應2.5h,得到淡藍色半透明溶液,即為所制備的鋁摻雜氧化鋅納米粒子溶液;
f)將步驟e)得到的淡藍色溶液進行離心,取離心管底部膠狀物質,待步驟e)中所得溶液離心完畢后,加入一定量的甲醇,超聲,使得納米粒子完全分散在甲醇中,離心,取底部膠狀物質。
g)將步驟f)所得膠狀物質用有機溶液分散,最終所得納米粒子分散液2ml,濃度為20mg/ml,最后向其中加入乙二胺(eda),向其中加入2μl乙二胺,最終所得產物淡藍色透明狀液體為本發明的azo納米粒子分散液。
實施例4。
a)稱取6.15g二水合醋酸鋅[zn(ch3coo)2·2h2o]、0.2g九水合硝酸鋁[al(no3)3·9h2o]加入氮氣瓶中,并加入275ml甲醇將其溶解;
b)稱取2.4g氫氧化鉀(koh)加入125ml甲醇中,超聲將其溶解;
c)在氮氣保護下,將步驟a)中所得溶液加熱到60℃,劇烈攪拌;
d)將步驟b)中所得溶液裝入恒壓滴液漏斗中,60℃下,將koh(aq)逐滴滴入步驟a)中所得溶液中,約45min滴完;
e)將步驟d)中所得混合溶液在80℃回流反應3.0h,得到淡藍色半透明溶液,即為所制備的鋁摻雜氧化鋅納米粒子溶液;
f)將步驟e)得到的淡藍色溶液進行離心,取離心管底部膠狀物質,待步驟e)中所得溶液離心完畢后,加入一定量的甲醇,超聲,使得納米粒子完全分散在甲醇中,離心,取底部膠狀物質。
g)將步驟f)所得膠狀物質用醇溶液分散,最終所得納米粒子分散液10ml,濃度為20mg/ml,最后向其中加入二乙烯三胺(deta),向其中加入10μl二乙烯三胺,最終所得產物淡藍色透明狀液體為本發明的azo納米粒子分散液。
實施例5。
a)稱取1.23g二水合醋酸鋅[zn(ch3coo)2·2h2o]、0.04g九水合硝酸鋁[al(no3)3·9h2o]加入氮氣瓶中,并加入55ml甲醇將其溶解;
b)稱取0.48g氫氧化鉀(koh)加入25ml甲醇中,超聲將其溶解;
c)在氮氣保護下,將步驟a)中所得溶液加熱到60℃,劇烈攪拌;
d)將步驟b)中所得溶液裝入恒壓滴液漏斗中,60℃下,將koh(aq)逐滴滴入步驟a)中所得溶液中,約20min滴完;
e)將步驟d)中所得混合溶液在80℃回流反應2.5h,得到淡藍色半透明溶液,即為所制備的鋁摻雜氧化鋅納米粒子溶液;
f)將步驟e)得到的淡藍色溶液進行離心,取離心管底部膠狀物質,待步驟e)中所得溶液離心完畢后,加入一定量的甲醇,超聲,使得納米粒子完全分散在甲醇中,離心,取底部膠狀物質。
g)將步驟f)所得膠狀物質用醇溶液分散,最終所得納米粒子分散液2ml,濃度為20mg/ml,最后向其中加入三乙烯四胺(teta),向其中加入2μl三乙烯四胺,最終所得產物淡藍色透明狀液體為本發明的azo納米粒子分散液。
上述實施例中所制得的樣品經儀器檢測進行表征,其結果如下:
1.由圖1可知,久置的azo溶液穩定性很好,沒有出現沉淀。
2.由圖2可知,所制備的azo沒有改變原始zno的晶體結構,均為纖鋅礦結構,且無雜峰出現。
3.由圖3可知,加入表面活性劑后的azo尺寸為10nm左右,與未加表面活性劑的azo(25nm)相比尺寸有所減小,并且團聚現象也得到改善,有利于形成更加均勻的薄膜。
4.由圖4、圖5、圖6、圖7、圖8對比可知,與未加入表面活性劑的azo薄膜(圖4)相比,加入表面活性劑之后azo薄膜(圖5、圖6、圖7、圖8)電流的強度和均勻性都有所提高,導電性的提高有利于制備厚度不敏感的界面層,對于實現制備柔性可拉伸器件和卷對卷大面積印刷具有重要意義。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種鋁摻雜氧化鋅納米粉體及其微波輔助制備方法與應用
- B摻雜SiC納米線在場發射陰極材料中的應用
- 一種基于負載型雙金屬共摻雜納米復合材料的電化學傳感器的制備方法
- 一種楔形Sn摻雜氧化鐵納米棒的制備方法
- 一種汽車電子封裝用納米氧化釔摻雜改性鋁鎂合金材料及其制備方法
- 具有鐵磁性B摻雜g-C<sub>3</sub>N<sub>4</sub>二維超薄納米片的制備方法
- 基于蛋白生物模板的釓摻雜硫化銅納米顆粒及其制備方法
- 制備Bi<sub>4</sub>V<sub>2</sub>O<sub>11</sub>納米晶及銀摻雜的BIAGVOX1.0 納米晶的方法
- 一種可摻雜的鉬酸鍶納米材料制備方法
- 一種柔性鋁摻雜氧化鋅/石墨烯復合材料的制備工藝的制作方法
- 一種氧摻雜碳化氮?貴金屬復合光催化劑、制備方法及應用與流程
- 一種摻銦氧化鋅粉體摻雜塑膠專用條狀鋁顏料的制作方法與工藝
- 一種多孔鋅摻雜四氧化三鐵/聚苯胺復合材料的制作方法與工藝
- 水熱結合鋅和氟復合摻雜制備具有優良循環及倍率性能的錳酸鋰正極材料的方法與流程
- 一種Ni,Sb?雙摻雜四氧化三鈷納米氧化物的制備方法與流程
- 一種建立仿真模型驗證氧化鋅摻雜鈀對氣敏性影響的方法與流程
- 一種Ho3+/Yb3+/Gd3+共摻雜氧化鋅上轉換發光材料及制備方法與流程
- 一種Ho3+/Yb3+/Li+共摻雜氧化鋅上轉換發光材料及制備方法與流程
- 一種W與Eu共摻雜的二氧化釩薄膜及其制備方法與流程
- 一種摻鋁氧化鋅(AZO)中空納米纖維的制備方法與流程