一種三維石墨烯結構體/高質量石墨烯的制備方法與流程
本發明屬于材料技術領域,涉及三維石墨烯結構體/高質量石墨烯的制備方法,具體為以氧化劑和酸為插層劑,得到石墨層間化合物;將所得的石墨層間化合物在膨脹劑中進行膨脹,得到三維石墨烯結構體;將所得三維結構體進行砂磨、球磨、剪切、超聲和高速流體粉碎等物理機械作用,得到石墨烯分散液。
背景技術:
碳元素由于其獨特的電子結構,能通過不同的雜化方式形成種類繁多的物質。其中,以sp2雜化方式存在的各類碳納米材料(包括富勒烯、碳納米管、石墨烯等)由于優異的電學、力學、熱學等性質,已成為材料領域的研究熱點。為充分發揮碳材料的優勢,開發出具有高比表面積的碳材料至關重要。高比表面積碳材料具有更多暴露的反應活性位點,豐富的空隙結構,能夠提高碳材料在應用過程中的各種性能。例如,將高比表面積碳材料應用于超級電容器、鋰離子電池等領域時,其單位體積或單位質量存儲的電量相比塊狀碳材料具有顯著優勢。
自石墨烯在2004年采用黏膠帶技術被成功制備以來,以石墨烯納米片層為構造單元制備出三維石墨烯材料被各國研究者廣泛研究。碳納米片層能夠減小離子在溶液中的擴散距離,經組裝后具有豐富的空隙結構,在儲能、催化等領域被廣泛應用。以介孔硅、氧化鈣等為模板劑,通過化學氣相沉積的方法在模板劑孔洞中生長出石墨烯納米片,將模板劑去除后,可得到三維高比表面積石墨烯材料。但這些方法成本高,路線冗長而難以工業化。氧化石墨烯是最為常見的制備三維高比表面積石墨烯材料的前驅體。氧化石墨烯由石墨為原料,經化學氧化后通過超聲、剪切或震蕩等方式分散在水或極性有機溶劑中。氧化石墨烯水溶液在一定濃度下,通過水熱或化學還原組裝,可以制得高比表面積的三維石墨烯結構體。此外,在氧化石墨烯溶液還原過程中不加攪拌等擾動,通過靜置還原的方式,也可以制得此類三維結構體。然而,由氧化還原法得到的石墨烯材料片層上有較多的缺陷,降低的力學、電學和熱學性能影響其實際應用,比如在儲能材料中,循環穩定性會因此降低。因而,開發以碳納米片層為構造單元的高比表面積、且晶格結構完整的石墨烯三維結構材料至關重要。
技術實現要素:
本發明的目的在于克服上述困難,提供一種三維石墨烯結構體/高質量石墨烯的制備方法。本發明方法制備的石墨烯三維結構體具有超過1000 m2/g的高比表面積,高于以氧化石墨烯為原料組裝得到的三維石墨烯結構體,并且石墨烯晶格結構完整,保留了碳材料優異的物理化學性質。將制備的三維結構體進行砂磨、球磨、剪切、高速流體粉碎或超聲等機械處理,可形成穩定的石墨烯分散液。本發明制備的石墨烯材料能應用于儲能、催化、復合材料等領域,具有操作流程簡單、成本低、可控性好等優勢,適合大規模生產和工業化應用。
本發明提出的一種三維石墨烯結構體/高質量石墨烯的制備方法,具體步驟如下:
(1)將原料石墨在插層劑中進行插層處理,使插層劑進入石墨層間,制得石墨層間化合物;
(2)將步驟(1)得到的石墨層間化合物投入到膨脹劑中,攪拌十五分鐘后靜置1-48h,使膨脹劑充分進入石墨層間,并在層間分解放出氣體,增大石墨烯的層間距,使得石墨烯片層彼此分離,形成具有超高比表面積的三維石墨烯結構體;
(3)將步驟(2)得到的三維石墨烯結構體在處理劑中通過超聲、砂磨、球磨、高速流體粉碎或剪切中的任何一種方式或它們的聯合方式處理后,得到石墨烯分散液。
本發明中,所述的原料石墨為天然鱗片石墨或人造石墨。
本發明中,步驟(1)中所述的插層劑由酸和氧化劑組成,所述酸為硫酸、磷酸或硝酸中任一種,所述氧化劑為硫酸、磷酸、硝酸、高錳酸鉀、高鐵酸鉀、重鉻酸鉀、三氧化鉻或雙氧水的一種或它們之間的組合。插層的目的在于削弱石墨片層間的范德華力,引入少量的含氧官能團。上述列出的插層劑種類可以選擇其中之一進行插層,也可以是它們之間的任意組合,但不限于此。
本發明中,步驟(1)中所述的氧化劑的加入量為原料石墨質量的0.1-20倍;酸的加入量為原料石墨質量的10-100倍。
本發明中,步驟(2)中所述的膨脹劑由酸和氧化劑組成,所述酸為硫酸或磷酸中任一種,所述氧化劑為三氧化硫、雙氧水、過硫酸銨、過硫酸鉀或過硫酸鈉中的一種或它們之間的組合。膨脹劑中,氧化劑和酸會進入石墨層間,氧化劑在層間分解放出氣體。氣體的產生克服了石墨層間的范德華力,使石墨發生膨脹。
本發明中,步驟(2)中所述氧化劑加入量為原料石墨的0.1-20倍,酸用量為原料石墨的10-200倍。
本發明中,步驟(3)中所述超聲時控制超聲功率為50-5000 W,超聲時間為 15 min-5 h;所述砂磨時控制砂磨機轉速為150-1600 rpm,砂磨時間為0.5-12h;所述球磨時控制球磨機轉速為150-1600 rpm,球磨時間為0.5-12h;所述高速流體粉碎時控制壓力為10-150 MPa,流速:300-40000 L/h;所述剪切時控制剪切速率為1000-30000 rpm,剪切時間為0.5-10h。
本發明中,所述處理劑采用N,N- 二甲基甲酰胺、 N- 甲基吡咯烷酮、鄰二氯苯、二甲基亞砜、甲苯、叔丁醇、異丙醇或乙醇與水的混合溶劑中的一種或幾種,或者是含有活性助劑的水溶液,活性助劑為聚乙烯吡咯烷酮、聚乙烯醇、聚乙二醇、直鏈烷基苯磺酸鈉、α-烯基磺酸鈉、脂肪醇聚氧乙烯醚硫酸鈉、十二烷基硫酸鈉、烷基酚聚氧乙烯醚、烷基醇酰胺、脂肪醇聚氧乙烯醚、十二烷基二甲基甜菜堿、十六烷基二甲基氯化銨、氫氧化鈉、非離子有機氟表面活性劑或聚醚改性聚有機硅氧烷等中任一種。
本發明中,制備得到的三維石墨烯結構體/高質量石墨烯比表面積可達1000 m2/g以上,石墨烯片層晶體結構保持完好,產率接近100%;所得三維石墨烯結構體通過砂磨、球磨、超聲、高速流體粉碎、剪切等作用,可實現完全剝離,在有機溶劑或表面活性劑水溶液中形成穩定的石墨烯分散液。所得石墨烯層數為1-10層,產率可達100%,石墨晶格完整度高,電導率可達1000 S/cm以上。
針對現有技術存在的不足,本發明人經過長期的實踐與研究,提出了本發明的技術方案,該方案可實現三維石墨烯結構體/高質量石墨烯的高效、低成本、大規模制備,解決了三維石墨烯結構體和高質量石墨烯分散液規模化制備的關鍵問題,為實現石墨烯在各個領域(如儲能材料、復合材料、催化、導電膜、散熱膜)中個實際應用提供了有效途徑。
本發明與現有技術相比具有以下優勢:
(1)以人造石墨或天然鱗片石墨為原料,原料來源廣泛,成本低廉。
(2)本發明操作條件溫和,不涉及石墨的氧化還原過程,不需要使用毒性較大的強還原劑,也不需要使用純氫氣在高溫下還原,避免了發生爆炸的危險。
(3)本發明無需將石墨完全氧化成為氧化石墨烯,再經組裝得到三維石墨烯,而是以石墨為原料,直接通過膨脹方式獲得,大大簡化了制備石墨烯三維結構體的流程,顯著提高了石墨烯三維結構體的質量。本發明制得的三維石墨烯結構體比表面積高,氧化程度低,有利于進一步表面修飾及與其它材料形成復合材料。
(4)本發明中石墨烯分散液制備方法具有產率高、質量高、能耗低的特點。
附圖說明
圖1為三維石墨烯結構體的掃描電鏡圖像。
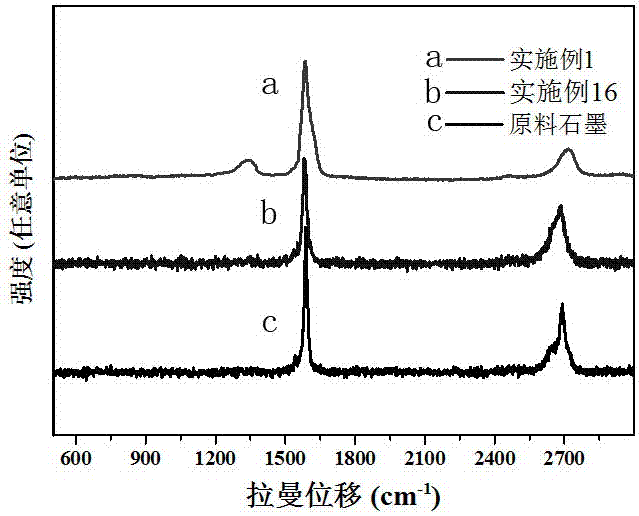
圖2為石墨、實施例1和16獲得的三維石墨烯結構體的拉曼譜。
具體實施方式
以下通過具體實例說明本發明的技術方案。應該理解,本發明提到的一個或多個步驟不排斥在所述組合步驟前后還存在其它方法和步驟,或者這些明確提及的步驟之間還可以插入其它方法和步驟。還應理解,這些實例僅用于說明本發明而不用于限制本發明的范圍。除非另有說明,各方法步驟的編號僅為鑒別各方法步驟的目的,而非為限制每個方法的排列次序或限定本發明的實施范圍,其相對關系的改變或調整,在無實質技術內容變更的條件下,當亦視為本發明可實施的范疇。
實施例1
(1)將90毫升濃硫酸(98%)在冰水浴條件下緩慢加入30毫升濃硝酸(65%)中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,過濾除去反應液,所得固體即為石墨層間化合物。
(2)將240毫升濃硫酸(98%)在冰水浴條件下緩慢加入60毫升雙氧水(30%)中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹48小時,過濾、水洗,得到三維石墨烯結構體,比表面積1360 m2/g,產率100%。圖1給出了掃描電子顯微鏡結果,圖2是相應拉曼譜。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL N-甲基吡咯烷酮中,用水浴超聲 30 min,得到黑色懸浮液,其中含有單層和少層石墨烯,剝離產率約100%。
實施例2
(1)將660毫升濃硫酸(98%)在冰水浴條件下緩慢加入220毫升濃硝酸(65%)中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,過濾除去反應液,所得固體即為石墨層間化合物。
(2)將240毫升濃硫酸(98%)在冰水浴條件下緩慢加入60毫升雙氧水(30%)中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹24小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1250 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL N-甲基吡咯烷酮中以35%輸出功率探頭超聲 30 min,得到單層和少層石墨烯,產率約100%。
實施例3
(1)將90毫升濃硫酸(98%)在冰水浴條件下緩慢加入30毫升濃硝酸(65%)中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑500微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將540毫升濃硫酸(98%)在冰水浴條件下緩慢加入135毫升雙氧水(30%)中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1320 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL N-甲基吡咯烷酮中在18000 rpm條件下剪切1h,得到單層和少層石墨烯,產率約100%。
實施例4
(1)在冰水浴條件下,將90毫升濃硫酸(98%)緩慢加入30毫升濃硝酸(65%)中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑500微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入300毫升硫酸中,并將步驟(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹1小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體。比表面積1250 m2/g,產率100%。
(3)將濕態三維石墨烯結構體用N-甲基吡咯烷酮將水置換出來,抽濾后的濕態物料置于球磨罐中,300 rpm條件下球磨5h。向球磨后的物料中加入N-甲基吡咯烷酮,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 N-甲基吡咯烷酮,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例5
(1)在冰水浴條件下,將90毫升硫酸緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入20毫升硫酸中,并將步驟(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體。比表面積1300 m2/g,產率100%。
(3)將濕態三維石墨烯結構體用N-甲基吡咯烷酮將水置換出來,抽濾后的濕態物料置于球磨罐中,500 rpm條件下砂磨6h。向砂磨后的物料中加入N-甲基吡咯烷酮,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 N-甲基吡咯烷酮,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色
實施例6
(1)將90毫升硫酸在冰水浴條件下緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將240毫升硫酸在冰水浴條件下緩慢加入60毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,過濾、水洗,得到三維石墨烯結構體。比表面積1320 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于100 mL 乙醇和水混合液中,水浴超聲 30 min,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入乙醇和水混合液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例7
(1)將660毫升硫酸在冰水浴條件下緩慢加入220毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將240毫升硫酸在冰水浴條件下緩慢加入60毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1230 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL乙醇和水混合液中以35%輸出功率探頭超聲 30 min,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入乙醇和水混合液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例8
(1)將90毫升硫酸在冰水浴條件下緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將540毫升硫酸在冰水浴條件下緩慢加入135毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1050 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL乙醇和水混合液液中,在18000 rpm條件下剪切1h,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入乙醇和水混合液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例9
(1)在冰水浴條件下,將90毫升硫酸緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入20毫升硫酸中,并將(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體。比表面積1250 m2/g,產率100%。
(3)向濕態三維石墨烯結構體在300 rpm條件下球磨5h。向球磨后的物料中加入乙醇和水混合液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 乙醇和水混合液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例9
(1)在冰水浴條件下,將90毫升硫酸緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入20毫升硫酸中,并將步驟(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體,比表面積1100 m2/g,產率100%。
(3)向濕態三維石墨烯結構體在800 rpm條件下砂磨2h。向砂磨后的物料中加入乙醇和水混合液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入乙醇和水混合液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例11
(1)將90毫升硫酸在冰水浴條件下緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將240毫升硫酸在冰水浴條件下緩慢加入60毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,過濾、水洗,得到三維石墨烯結構體。比表面積1200 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于100 mL 0.5 mg/mL聚乙烯吡咯烷酮水溶液中,水浴超聲 30 min,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例12
(1)將660毫升硫酸在冰水浴條件下緩慢加入220毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將240毫升硫酸在冰水浴條件下緩慢加入60毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1250 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL0.5 mg/mL聚乙烯吡咯烷酮水溶液中以35%輸出功率探頭超聲 30 min,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例13
(1)將90毫升硫酸在冰水浴條件下緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),并在室溫條件下攪拌24小時。隨后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將540毫升硫酸在冰水浴條件下緩慢加入135毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1150 m2/g,產率100%。
(3)將0.1g濕態三維石墨烯結構體置于 100 mL 0.5 mg/mL聚乙烯吡咯烷酮水溶液中,在18000 rpm條件下剪切1h,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例14
(1)在冰水浴條件下,將90毫升硫酸緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入20毫升硫酸中,并將步驟(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體。比表面積1010 m2/g以上。
(3)向濕態三維石墨烯結構體中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,300 rpm條件下球磨5h。向球磨后的物料中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例15
(1)在冰水浴條件下,將90毫升硫酸緩慢加入30毫升硝酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入20毫升硫酸中,并將步驟(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體。比表面積910 m2/g以上。
(3)向濕態三維石墨烯結構體中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,600 rpm條件下砂磨4h。向砂磨后的物料中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例16
(1)在冰水浴條件下,將3克重鉻酸鉀緩慢加入100毫升濃硫酸中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將0.3克過硫酸銨加入20毫升硫酸中,并將步驟(1)中的石墨層間化合物加入混合液中,35℃攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到三維石墨烯結構體。比表面積1020 m2/g。圖2給出了相應的拉曼譜。
(3)向濕態三維石墨烯結構體中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,600 rpm條件下砂磨4h。向砂磨后的物料中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例17
(1)在冰水浴條件下,將18克三氧化鉻緩慢加入100毫升硫酸,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將240毫升硫酸在冰水浴條件下緩慢加入60毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹10小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1160 m2/g,產率100%。
(3)向濕態三維石墨烯結構體中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,600 rpm條件下砂磨4h。向砂磨后的物料中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
實施例18
(1)在冰水浴條件下,將3克高錳酸鉀緩慢加入80毫升濃硫酸和20毫升濃磷酸混合液中,混合均勻后,向反應液中加入3克鱗片石墨(顆粒直徑150微米,含碳量高于95%),在室溫條件下攪拌24小時后,將反應液用砂芯漏斗抽濾,所得固體即為石墨層間化合物。
(2)將250毫升硫酸在冰水浴條件下緩慢加入50毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將步驟(1)中的石墨層間化合物加入混合液中,室溫攪拌15分鐘后靜置膨脹6小時,洗滌、過濾,得到三維石墨烯結構體。比表面積1210 m2/g,產率100%。
(3)向濕態三維石墨烯結構體中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,300 rpm條件下球磨5h。向球磨后的物料中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
對比例1
(1)將240毫升硫酸在冰水浴條件下緩慢加入60毫升雙氧水中,冷卻后將混合液移入40℃水浴中。隨后,將1克原料石墨(顆粒直徑150微米,含碳量高于95%)加入混合液中,室溫攪拌15分鐘后靜置膨脹3小時,洗滌、過濾,直至濾液為中性,得到產物。石墨發生膨脹,比表面積241 m2/g。
(2)將0.1g(1)中濕態產物體置于100 mL N-甲基吡咯烷酮,水浴超聲 30 min,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入N-甲基吡咯烷酮,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
對比例2
(1)將30克過硫酸鈉加入300毫升硫酸中,并將3克原料石墨(顆粒直徑150微米,含碳量高于95%)加入混合液中,35℃攪拌15分鐘后靜置膨脹6小時,洗滌、過濾,直至濾液為中性,得到產物。石墨發生膨脹,比表面積60 m2/g。
(2)將0.1g步驟(1)中濕態產物體置于100 mL N-甲基吡咯烷酮,35%輸出功率下探頭超聲 30 min,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入N-甲基吡咯烷酮,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
對比例3
(1)將30克過硫酸鉀加入300毫升硫酸中,并將3克原料石墨(顆粒直徑150微米,含碳量高于95%)加入混合液中,35℃攪拌15分鐘后靜置膨脹6小時,洗滌、過濾,直至濾液為中性,得到產物。石墨發生膨脹,比表面積64 m2/g。
(2)將步驟(1)中產物用N-甲基吡咯烷酮(1)中產物內部的水置換出來,抽濾后的濕態物料置于球磨罐中,300 rpm條件下球磨5h。向球磨后的物料中加入N-甲基吡咯烷酮,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 N-甲基吡咯烷酮,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
對比例4
(1)將30克過硫酸銨加入300毫升硫酸中,并將3克原料石墨(顆粒直徑150微米,含碳量高于95%)加入混合液中,35℃攪拌15分鐘后靜置膨脹6小時,洗滌、過濾,直至濾液為中性,得到產物。石墨發生膨脹,比表面積67 m2/g。
(2)向0.1g步驟(1)中濕態產物加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,600 rpm條件下砂磨4h。向砂磨后的物料中加入0.5 mg/mL聚乙烯吡咯烷酮水溶液,得到黑色懸浮液,在 12000 rpm下離心15 min,倒去上清液,再加入 0.5 mg/mL聚乙烯吡咯烷酮水溶液,在 1000 rpm下離心 30 min,重復離心并收集上層液,直至上層液為無色。
- 還沒有人留言評論。精彩留言會獲得點贊!