一種具有分層微結構的硫化錳微球及其制備方法與流程
本發明涉及一種具有分層微結構的硫化錳微球及其制備方法。
背景技術:
在納米技術領域,通過自主裝法合成三維(3D)分層納米微結構進行了許多努力,其預期能夠顯示一些新的性質。順磁材料中的硫化錳由于其獨特的電,光學和磁性已被用于許多領域。MnS晶體有三個相,包括穩定的α-MnS和兩個亞穩相β-MnS和γ-MnS,相比于α-MnS,β-MnS和γ-MnS,顯示出更為優越的性能;過去幾年,在控制硫化錳的晶型結構方面做了許多努力。然而,合成亞穩相的硫化錳仍然非常困難,因為其不具有熱穩定性,并且在高溫或高壓的條件下容易發生從亞穩相到穩定相的轉變。此外,由于納米材料的物理和化學性質不僅取決于相,而且還取決于尺寸,形狀和結構,因此合成具有受控形態的MnS晶體已經受到了極大的關注;迄今為止,已經成功地制備了具有各種尺寸和形狀的MnS納米晶體,例如棒,立方體,星形,珊瑚,花,雙腳,蒲公英,等等。在這些形態中,與其固體對應物相比,3D分層納米/微結構可提供一種新的方式來探索它們的新性質(特別是磁性)。例如,由于樣品的小尺寸和自組裝性質,MnS超結構顯示出大的矯頑場(例如,40nm納米管組件的1265Oe),然而,納米粒子自組裝法合成MnS超結構的方法依然很少。采用簡易快速方法制備具有受控形態的超結構MnS仍然是一個巨大的挑戰。在這里,我們研究了一個簡易的水熱方法,合成了納米粒子組裝的可控分層微米結構MnS。此外,發現MnS的形態對其磁性能施加顯著的影響。
技術實現要素:
本發明的目的在于發明一種具有分層微結構的硫化錳微球及其制備方法;本發明是這樣實現的,根據本發明的一個方面,一種具有分層微結構的硫化錳微球的制備方法,首先將醋酸錳和尿素以3:1~5:1的比例加入到10-20mL的L-半胱氨酸水溶液中,室溫攪拌10~15min;再將上述混合溶液倒入容積為30~50mL的高壓反應釜中,180℃條件下反應10h,冷卻至室溫,將所述混合溶液離心分離,將得到的固體分別用蒸餾水和酒精洗滌三次;最后再將洗滌后的固體在干燥箱中干燥10~12h;L-半胱氨酸水溶液的濃度為0.08~0.12mol/L,當所述L-半胱氨酸水溶液的濃度為0.08mol/L時,合成的硫化錳微球是大量均勻且組織良好的直徑為6.0-8.3μm的分層微球,由直徑為20-25nm的納米顆粒構成;所述L-半胱氨酸水溶液的濃度為0.09mol/L時,硫化錳微球是均勻的微球體,直徑在6.2-8.8μm的范圍內,并且所述微球體由直徑為150-200nm的納米顆粒構成。當L-半胱氨酸水溶液的濃度為0.10mol/L,為直徑為6.3-8.4μm的微球,所述微球由直徑為250-400nm的納米顆粒構成;所述L-半胱氨酸水溶液的濃度為0.12mol/L時,硫化錳微球是直徑為7.3-9.4μm的微球體,微球由直徑為500-700nm納米顆粒構成。上述制備的球形MnS層級具有超順磁性,使得MnS顆粒在施加磁場下易于定向和分離,并且在生物醫學領域具有潛在的應用。
通常,晶體形態由其晶體結構決定。因此,在L-半胱氨酸的高濃度下,易形成具有大尺寸的六邊形納米盤。簡而言之,L-半胱氨酸不僅作為硫源來以控制核速率和生長速率,而且也作為穩定劑來控制MnS的晶體尺寸和形態。
附圖說明
圖1為實施例1制備的具有分層微結構的硫化錳微球的晶體結構;
大球代表S,小球代表Mn;
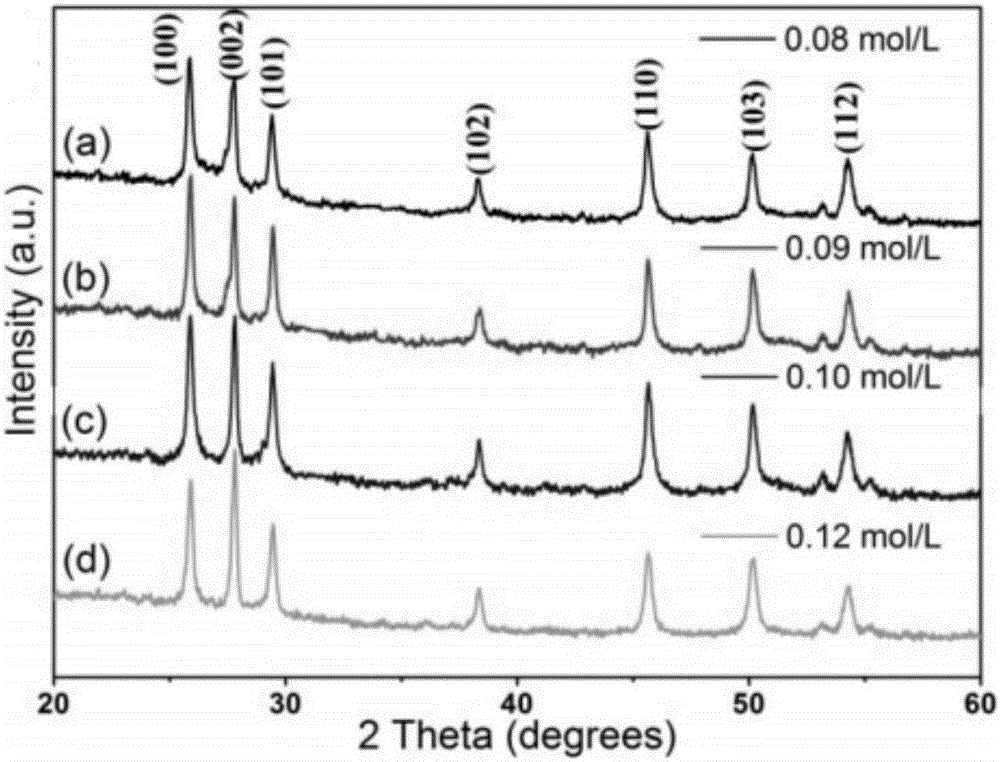
圖2為實施例1制備的具有分層微結構的硫化錳微球的XRD圖;
(a)L-半胱氨酸的濃度為0.08mol/L;(b)L-半胱氨酸的濃度為0.09mol/L;(c) L-半胱氨酸的濃度為0.10mol/L;(d)L-半胱氨酸的濃度為0.12mol/L。
圖3為實施例1制備的具有分層微結構的硫化錳微球的XPS圖;
(A)總譜圖(B)Mn 2p的分峰譜圖(C)S 2p的分峰譜圖
圖4為實施例1制備的具有分層微結構的硫化錳微球的SEM圖;
a1,a2,a3,a4為L-半胱氨酸的濃度為0.08mol/L時
b1,b2,b3,b4為L-半胱氨酸的濃度為0.09mol/L時
c1,c2,c3c4為L-半胱氨酸的濃度為0.10mol/L時
d1,d2,d3,d4為L-半胱氨酸的濃度為0.12mol/L時
圖5為實施例1制備的具有分層微結構的硫化錳微球的形成過程示意圖;
圖6為實施例1制備的具有分層微結構的硫化錳微球的初步磁滯回線的分析。
具體實施方式
下面結合附圖及實施方式對本發明作進一步詳細的說明:
應理解,這些實施例僅用于說明本發明而不用于限制本發明的范圍。
實施例1
制備具有分層微結構的硫化錳微球:1mmol醋酸錳和0.2mmol尿素加入到15mL含有不同濃度L-半胱氨酸(0.08,0.09,0.10and 0.12mol/L)的蒸餾水中,室溫攪拌15min。然后將溶液倒入30mL的高壓反應釜中,180℃反應10h。冷卻到室溫,離心分離,用蒸餾水和酒精沖洗三次,60℃干燥10h;
對上述制備的具有分層微結構的硫化錳微球,進行表征,圖1為γ-MnS的晶形結構圖,如圖2所示,從XRD圖中可以看出沒有其他的雜峰出現,表明樣品具有高純度,從圖中也可以觀察到隨著L-半胱氨酸濃度的升高,(002)和(100)的峰強度也逐漸增大,這可能是由于(002)的晶面暴露增大導致的,另一個現象是當L-半胱氨酸的濃度增大到0.1M時,(002)衍射峰明顯變窄,這是由于硫化錳晶型增大所導致的。然而,所有的XRD圖都是相似的,說明L-半胱氨酸的量對硫化錳晶型結構無影響。
如圖3所示為硫化錳微球的XPS圖,XPS a中可以發現C、O、Mn、S的特征峰,其中C的特征峰歸屬于支撐膜上的C,并且O峰歸因于樣品表面或支撐膜上的氧。b 638.6-660eV的特征峰歸屬于Mn 2p1/2和Mn 2p3/2位于640.9和642.3eV的峰都歸因于Mn 2p 3/2中氧化的Mn(Mn2+),同時,在653.4eV處只有一個峰也代表Mn 2p1/2中氧化的Mn(Mn 2+),C中160.8,161.7,163.2和164.5eV歸屬于S 2p;XPS分析表明,Mn和S的原子比約為1:1,這與整體MnS的化學計量一致。這些XPS結果與圖1所示的XRD數據具有良好的一致性,這表明合成的γ-MnS具有很高的純度。
圖4為當L-半胱氨酸的濃度為0.08mol/L的SEM圖,SEM圖像a1顯示大量直徑為6.0-8.3μm的均勻且組織良好的分層微球,高放大倍數的SEM圖像(圖4a2和4a3)顯示了微球結構的詳細信息,其由直徑為20-25nm的納米顆粒構成,當L-半胱氨酸的濃度增加到0.09mol/L時,產物仍顯示均勻的微球體,并且直徑在6.2-8.8μm的范圍內(圖4b1),其由直徑為150-200nm的納米顆粒構成(圖4b2和4b3)。
當L-半胱氨酸的濃度為0.10mol/L時,形態為直徑為6.3-8.4μm的球形形狀(圖4c1),由直徑為250-400nm的納米顆粒構成(圖4c2和4c3)。
進一步增加L-半胱氨酸的濃度至0.12mol/L,產物仍顯示均勻的微球體,直徑為7.3-9.4μm(圖4d1),但這些組分納米顆粒的尺寸顯示明顯增加,約500-700nm(圖4d2和4d3)。并且,這些組分納米顆粒的形態表現出六角形板,這與γ-MnS六方晶體結構相一致,并且也符合上述XRD結果。這些SEM結果表明MnS納米晶體的形態可以通過調節L-半胱氨酸的濃度來控制。也就是說,隨著使用較高濃度的L-半胱氨酸,MnS納米顆粒的晶體尺寸增加。
基于上述實驗結果,提出了一種MnS分級微球的反應機理,如圖5所示。首先,L-半胱氨酸水解和釋放H2S,H2S容易電離出S2-離子。然后,S2-離子可以直接與Mn2+離子反應并形成MnS團簇,做為晶體進一步生長的基體。加入低濃度的L-半胱氨酸(例如0.08mol/L),成核速率大于生長速率,因此形成小的納米晶體。然后,由于整體表面能的最小化,納米晶體具有自組裝形成MnS微球的傾向。這些微球經歷重構或重排成更緊密和致密的微球結構,如圖3a所示。在L-半胱氨酸的高濃度(例如0.10mol/L)的條件下,高濃度的S2-離子使生長速率大于成核速率,這將有助于形成更大的晶體形成。此外,在該生長過程中,多余的L-半胱氨酸分子將容易地吸附在生長的晶體的晶面上,并起到封端劑的作用以保護晶面。XRD結果(圖2)顯示所制備的MnS晶體是六方晶體結構。通常,晶體形態由其晶體結構決定。因此,在L-半胱氨酸的高濃度下,易形成具有大尺寸的六邊形納米盤。簡而言之,L-半胱氨酸不僅作為硫源來以控制核速率和生長速率,而且也作為穩定劑來控制MnS的晶體尺寸和形態。
為了研究形態如何影響MnS的磁性能,對上述制備的MnS樣品在室溫下進行初步磁滯回線的分析(圖6)。顯然,所有樣品中均具有較大的飽和磁化強度,并表現出順磁性,剩余磁化強度非常小,導致容易的磁消磁和磁性納米顆粒快速分散。此外,可以看出,滯回線的斜率隨著L-半胱氨酸添加量的增加而逐漸減小,這可能是由于MnS納米晶體的尺寸增加所導致的。因此,上述制備的球形MnS層級具有超順磁性,使得MnS顆粒在施加磁場下易于定向和分離,并且在生物醫學領域具有潛在的應用。
- 還沒有人留言評論。精彩留言會獲得點贊!