一種超聲輔助制備鎳鈰固溶體材料的方法與流程
本發明屬于固體材料制備技術領域,具體涉及一種制備鎳鈰固溶體材料的方法。
背景技術:
氧化鈰作為一種典型的稀土氧化物,其螢石晶體結構中同時存在+3價和+4價鈰離子,使其具有優異的氧化還原性能以及儲放氧性能,因此以氧化鈰為基礎的鈰基復合金屬材料被廣泛應用于環境催化和能源催化領域,如汽車尾氣催化氧化,揮發性有機物(VOCs)催化氧化,水蒸氣重整等。其中,將金屬離子摻雜到氧化鈰晶格中形成的鈰基固溶體材料因具有更高的氧空位含量、更快的離子遷移速率和更高的催化反應活性而受到廣泛關注。
研究發現,鈰基固溶體材料的性能主要受晶粒大小、晶相組成以及摻雜金屬離子種類等因素影響。目前,制備鈰基固溶體材料的方法主要有:共沉淀法、溶膠–凝膠法、化學燃燒法、微乳液法和水熱晶化法等。共沉淀法因工藝簡單,容易操作而被廣泛應用于工業生產中,但其制備的鈰基固溶體材料晶粒較大,晶相分布不均勻,導致催化活性較低。利用化學燃燒法、微乳液法等方法制備出的固溶體晶粒可以達到幾個納米大小,其比表面積和空穴相對含量明顯增多,具有較好的催化性能。例如,李燦(李燦,Appl. Catal. A: Gen., 2003,246,1)利用檸檬酸燃燒法制備了納米鎳鈰固溶體材料,并且發現當鎳鈰比為3 : 7時,催化劑具有最佳的氧化還原性能。Laguna等(Laguna,Appl. Catal. B: Environ., 2011, 106, 621)利用微乳液法制備了納米鐵鈰固溶體材料,其鐵含量可高達50at.%,并指出當鐵含量為10at.%時催化劑的氧空位濃度達到最高同時具有最高的氧化還原效率。
納米鈰基固溶體材料的制備在很大程度上改善了氧化鈰基催化劑的性能,但是上述幾種方法在制備過程中容易出現晶相分布不均勻,導致摻雜效率較低,同時影響催化劑的整體性能。因此,開發一種制備晶相均一的納米鈰基固溶體材料的方法對進一步優化鈰基催化劑材料性能具有非常重要的意義。
技術實現要素:
為了克服現有技術所存在的問題,本發明提供一種晶相均一的高性能鎳鈰固溶體的制備方法,本發明提供的高性能鎳鈰固溶體的制備方法,首次采用有機高分子作為結構導向劑和超聲輔助技術,利用高溫老化過程制備鎳鈰固溶體材料;其中,通過調節金屬鹽的比例、溶液的溫度、超聲處理的時間、模板劑的種類及水熱處理的溫度及時間,使制備得到的鎳鈰固溶體材料納米顆粒堆積組成,呈花狀或球狀等,形貌均一且結構均勻,并具有大比表面積和高儲放氧量等特點,儲氧量達到400–600μmol O/g,在環境催化、固體燃料電池、傳感器等領域具有良好的應用前景。
本發明提供的超聲輔助制備鎳鈰固溶體材料的方法,具體步驟為:
(1)將無機鈰源與無機鎳源按一定比例溶于有機溶劑二甲基亞砜中,并加入一定量有機酸,攪拌至混合均勻;其中,鈰鎳摩爾比為99:1–4:1,優先鈰鎳摩爾比為50:1–4:1;
(2)再加入一定量的有機高分子作為結構導向劑,并進行超聲處理,使溶液混合均勻,之后加入一定量的稀氨水,并進行充分攪拌;
(3)將步驟(2)中所得溶液轉入水熱釜中進行高溫處理,之后經過過濾、洗滌、干燥、焙燒、酸洗處理,即制得鎳鈰固溶體材料。
在步驟(1)中,所述無機鈰源為六水硝酸鈰(Ce(NO3)3·6H2O)或七水氯化亞鈰(CeCl3·7H2O);所用無機鎳源為六水硝酸鎳(Ni(NO3)2·6H2O)或六水氯化鎳(NiCl2·6H2O);所述有機酸為甲酸或乙醇酸。
在步驟(2)中,所述高分子結構導向劑為聚乙烯吡咯烷酮(分子量為10000–800000)或十六烷基溴化銨;所述超聲處理過程為采用40kHz頻率超聲波對溶液進行處理,處理時間為0.5–4h,超聲時溶液溫度為30–60℃。所述氨水濃度為1.5–3wt%。
在步驟(3)中,所述老化處理過程中,老化溫度為120–180℃,老化時間為0.5–72h;所述洗滌過程為將過濾后所得固體分別用蒸餾水及無水乙醇洗滌2-3遍;所述干燥過程為將過濾后的固體置于80℃烘箱中干燥6-8h;所述焙燒過程為將所得固體樣品在空氣氣氛中進行焙燒,焙燒溫度為400–600℃,升溫速率為2-3 ℃/min,焙燒時間為3-5h;所述酸洗過程為將所得固體置于稀硝酸中浸泡洗滌,稀硝酸濃度為5–15wt%,浸泡時間為0.5–6h,之后過濾。
本發明的優點:
(1)所制備的鎳鈰固溶體晶相均一,鎳離子摻雜效率高,氧空位濃度高;
(2)所制備的鎳鈰固溶體晶粒尺度小,比表面積大;
(3)所制備的鎳鈰固溶體鎳含量較低,金屬鎳摻雜效率高,原料廉價易得,制造成本較低。
附圖說明
圖1為實施例1~3及對比例1中所得鎳鈰固溶體的掃面電子顯微鏡(SEM)照片。
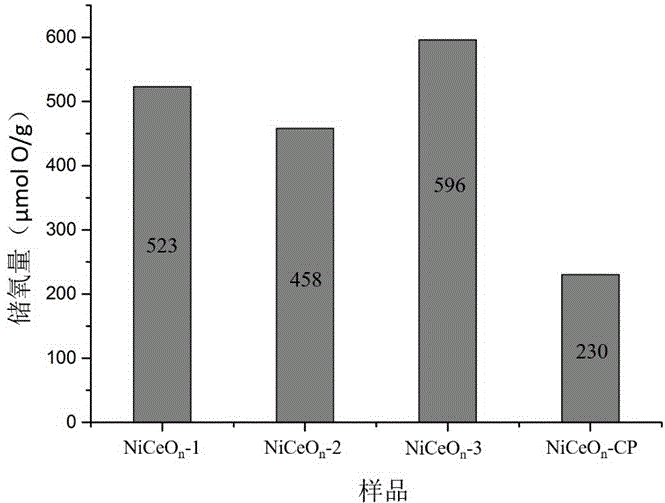
圖2為實施例1~3及對比例1中所得鎳鈰固溶體的儲氧量測試結果。
具體實施方式
下面將通過實例對于本發明做進一步的詳細說明,但并不因此對其有所限制。
實施例1
將0.181g七水氯化鈰和0.029g六水氯化鎳(摩爾比為4:1)加入到含有40ml二甲基亞砜的燒杯中,并加入1.0ml甲酸,攪拌30min后,將燒杯放入頻率為40kHz超聲清洗機中,同時緩慢加入1.0g聚乙烯吡咯烷酮(分子量為10000),設置超聲溫度為30℃,開始超聲處理,處理時間為0.5h。加入4ml濃度為1.5wt%的稀氨水并攪拌均勻,之后將溶液轉入配備聚四氟乙烯內襯的水熱釜中進行高溫處理,處理溫度為120℃,處理時間為0.5h。高溫處理結束后,過濾溶液,并分別用蒸餾水和無水乙醇洗滌固體3遍,之后在80℃下干燥6-8h,然后將固體在400℃空氣氣氛下進行焙燒5h,升溫速率2℃/min。所得固體浸泡于5wt%的稀硝酸溶液中處理0.5h,在經過上述相同的過濾、洗滌、干燥、焙燒過程,得到最終產物鎳鈰固溶體材料(記為NiCeOn-1),其形貌如圖1所示,其儲氧量如圖2所示。
實施例2
將2.321g六水硝酸鈰和0.016g六水硝酸鎳(摩爾比為99:1)加入到含有100ml二甲基亞砜的燒杯中,并加入3.0ml乙醇酸,攪拌30min后,將燒杯放入頻率為40kHz超聲清洗機中,同時緩慢加入4.0g十六烷基溴化銨,設置超聲溫度為60℃,開始超聲處理,處理時間為4h。加入8ml濃度為3wt%的稀氨水并攪拌均勻,之后將溶液轉入配備聚四氟乙烯內襯的水熱釜中進行高溫處理,處理溫度為180℃,處理時間為72h。高溫處理結束后,過濾溶液,并分別用蒸餾水和無水乙醇洗滌固體3遍,之后在80℃下干燥6-8h,然后將固體在600℃空氣氣氛下進行焙燒3h,升溫速率3℃/min。所得固體浸泡于15wt%的稀硝酸溶液中處理0.5h,在經過上述相同的過濾、洗滌、干燥、焙燒過程,得到最終產物鎳鈰固溶體材料(記為NiCeOn-2),其形貌如圖1所示,其儲氧量如圖2所示。
實施例3
將0.181g七水氯化鈰和0.013g六水氯化鎳(摩爾比為9:1)加入到含有34ml二甲基亞砜的燒杯中,并加入1.0ml甲酸,攪拌30min后,將燒杯放入頻率為40kHz超聲清洗機中,同時緩慢加入1.0g聚乙烯吡咯烷酮(分子量30000),設置超聲溫度為30℃,開始超聲處理,處理時間為1h。加入2ml濃度為2wt%的稀氨水并攪拌均勻,之后將溶液轉入配備聚四氟乙烯內襯的水熱釜中進行高溫處理,處理溫度為160℃,處理時間為4h。高溫處理結束后,過濾溶液,并分別用蒸餾水和無水乙醇洗滌固體3遍,之后在80℃下干燥6-8h,然后將固體在450℃空氣氣氛下進行焙燒4h,升溫速率2℃/min。所得固體浸泡于10wt%的稀硝酸溶液中處理2h,在經過上述相同的過濾、洗滌、干燥、焙燒過程,得到最終產物鎳鈰固溶體材料(記為NiCeOn-3),其形貌如圖1所示,其儲氧量如圖2所示。
對比例1
采用共沉淀法制備該對比例。將1.81g七水氯化鈰和0.13g六水氯化鎳(摩爾比為9:1)加入到含有50ml蒸餾水的燒杯中,充分溶解后,快速加入濃度為25wt%的氨水,調節溶液pH值到10左右,將燒杯放入50℃的水浴中繼續攪拌1h,之后轉入帶有聚四氟乙烯內襯的水熱釜中在120℃下高溫處理4h。高溫處理結束后,自然降溫,并進行過濾、洗滌、干燥及焙燒過程(處理方法同實施例3),得到共沉淀法制備的鎳鈰固溶體材料(記為NiCeOn-CP)。
- 還沒有人留言評論。精彩留言會獲得點贊!