一種氧化石墨烯的制備方法與流程
本發明屬于新材料制備技術領域,具體涉及一種氧化石墨烯的制備方法。
背景技術:
石墨烯是一層以六角形蜂巢結構周期性緊密堆積的碳原子構成的二維碳材料,是目前已知的密度最小、比表面積最大、載流子遷移率最大、楊氏模量最大、透光性最好、導電性能最好的材料,在儲能領域、電子領域、環保領域、復合材料領域、生物醫藥領域等方面擁有巨大的應用前景。但在實際應用中,也正是因為這些無與倫比的性能,石墨烯材料面臨著易團聚導致分散性差、界面相容性差、與其他材料難以融合的困擾。
氧化石墨烯作為一種表面功能化的石墨烯衍生物,由于其表面含有豐富的含氧官能基團,一方面削弱了石墨烯片層之間的相互作用力,賦予其優異的分散性能;另一方面提供了大量的化學反應活性點,易與其他材料復合形成良好的界面。該材料目前可通過兩種手段獲得:一是石墨經氧化插層處理后經熱膨脹或機械手段進行深度剝離,得到氧化石墨烯,二是以非氧化還原獲得的結構完整的石墨烯為原料,經氧化處理得到氧化石墨烯,其中前者為主流制備工藝,具體方法主要有Brodie法、Staudenmaier法、Hummers法和改進的Hummers法。
Brodie法在濃硝酸體系中添加KClO3為強氧化劑,其氧化程度可通過改變氧化時間來進行控制,合成的氧化石墨結構較為完整,但需要經過多次氧化,反應時間相對較長,且過程中會產生較多的ClO2氣體;Staudenmaier法是在濃硝酸和濃硫酸體系中以KClO3為氧化劑,反應過程中會產生較多的Cl2和ClO2等有毒氣體,且對氧化石墨結構的破壞程度較嚴重;Hummers法以其氧化時間短、氧化程度較高、產物結構較規整、安全系數較高等優點成為目前最常用的制備方法,但該方法采用KMnO4做為深度氧化劑,易導致產品淬滅后洗滌困難,Mn離子難于清洗脫除而導致產品純度不高;此外,氧化石墨烯片層結構出現部分重疊,石墨層間距小,難于剝離成單層結構,清洗不干凈甚至可能導致產品質量不達標,無法滿足要求,造成較大的經濟損失;改進的Hummers法減少了硝酸鹽的使用,能夠獲得具有高氧化程度的氧化石墨,但是該方法仍沒有避免雜質離子的引入、且后續多次洗滌除雜過程復雜,仍存在產品質量不達標的風險。
技術實現要素:
本發明所要解決的技術問題是,克服以上背景技術中提到的不足和缺陷,提供一種制備產品純度高、安全性好、產品片層結構間距大、廢酸可循環回用、制備過程綠色環保的氧化石墨烯的制備方法。
為解決上述技術問題,本發明提出的技術方案為一種氧化石墨烯的制備方法,包括以下步驟:
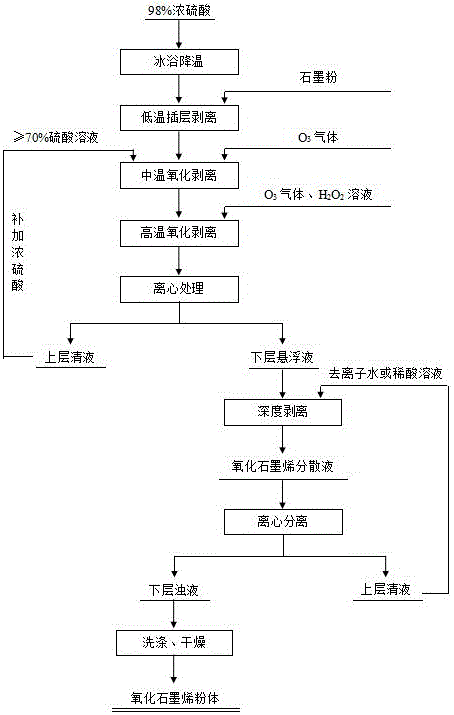
(1)將石墨粉加入到低溫下的濃硫酸中,在攪拌條件下同時輔以超聲處理以進行低溫插層剝離;
(2)在上述步驟(1)中進行的低溫插層剝離完成后,向所得的混合溶液中加入硫酸溶液,并持續通入含臭氧氣體作為氧化劑,然后逐步升溫到中溫環境進行恒溫攪拌反應,以實現中溫氧化剝離;
(3)在上述步驟(2)的中溫氧化剝離完成后,持續通入含臭氧氣體作為氧化劑,然后開始加入強氧化劑溶液,并逐步升溫到高溫環境進行恒溫攪拌反應,以實現高溫氧化剝離;所述步驟(2)和步驟(3)中的攪拌反應均優選是在超聲條件下進行;
(4)在上述步驟(3)的高溫氧化剝離完成后,停止通入含臭氧氣體,然后將所得的混合液進行離心處理,分離出上層清液(分離出的上層清液的體積一般為離心處理前混合溶液總體積的60%~85%)和下層懸浮液,對下層懸浮液進行深度剝離后得到含氧化石墨烯的分散液,或者對下層懸浮液進行深度剝離、離心分離、洗滌及干燥處理后得到氧化石墨烯粉體。
上述的氧化石墨烯的制備方法,優選的,所述石墨粉為天然鱗片石墨,其純度為≥99%,粒徑為40-1000目。
上述的氧化石墨烯的制備方法,優選的,所述低溫下的濃硫酸為-5℃-5℃的98%濃硫酸(如無特別說明,硫酸的百分濃度均是指質量百分比濃度)。
上述的氧化石墨烯的制備方法,優選的,所述步驟(1)中,石墨粉與濃硫酸的固液比控制為1:10~80 g/ml(即每克石墨粉配加10~80 ml的濃硫酸)。
上述的氧化石墨烯的制備方法,優選的,所述步驟(2)中,所述硫酸溶液的質量濃度≥70%,在添加所述硫酸溶液之后,使混合溶液中石墨粉的用量與硫酸溶液總的液固比控制在1:30~100 g/ml。
上述的氧化石墨烯的制備方法,優選的,所述步驟(2)和步驟(3)中,所述含臭氧氣體中O3濃度≥50%(其可以是空氣源O3、純氧源O3中的一種或兩種的組合,臭氧一般為O3發生器所制);所述含臭氧氣體的流速控制為0.1~10.0 m3/h。
上述的氧化石墨烯的制備方法,優選的,所述步驟(2)中,中溫環境是指30℃-50℃溫度條件。
上述的氧化石墨烯的制備方法,優選的,所述步驟(3)中,高溫環境是指60℃-80℃溫度條件;所述恒溫攪拌反應的時間至少在5min以上。
上述的氧化石墨烯的制備方法,優選的,所述步驟(3)中,所述強氧化劑溶液為質量分數30%的過氧化氫溶液,過氧化氫溶液的加入量按照混合溶液中每克石墨粉配加5~50ml的過氧化氫溶液計算。通過在O3氧化過程中加入適量的過氧化氫溶液,可起到促進自由基生成、進一步增強氧化作用的效果。
上述的氧化石墨烯的制備方法,優選的,所述步驟(2)中,所述硫酸溶液是源自步驟(4)中分離出的上層清液或者是源自前述上層清液與濃硫酸混合后得到的混合液。
上述的氧化石墨烯的制備方法,優選的,所述步驟(4)中,所述深度剝離是指向分離出的下層懸浮液中加入去離子水或稀酸溶液,在攪拌和超聲處理條件下使其深度反應5min~300min;所述稀酸溶液是由含氧化石墨烯的分散液進行深度剝離、離心分離后所得的上層清液來充當。
上述本發明的技術方案主要基于以下原理:
一是在低溫條件下,充分利用濃硫酸的化學插層作用來破壞石墨的邊緣結構并侵入結構內部,同時充分利用超聲波的機械剪切作用和空化作用來破壞原子間作用力從而增大石墨層間距,為后續石墨的充分氧化和層間剝離提供有利條件;
二是在低溫插層剝離初步破壞石墨結構的基礎上,借助攪拌和超聲波的雙重機械作用強化石墨的氧化剝離,充分利用O3分子的氣體流動性來加強石墨層間的傳質過程,使得自由基與石墨碳原子充分接觸,起到強化石墨層間的氧化作用和增大石墨層間距的雙重目的;
三是充分利用O3在酸性體系中的強氧化能力來實現石墨的深度氧化,一方面利用O3的直接氧化作用來破壞苯環結構中的碳碳雙鍵,為自由基的結合提供活化位點;另一方面O3分解過程中會產生氧化活性很高的自由基,充分利用該自由基的化學活性,使其在與石墨碳原子結合并將其氧化的同時,部分自由基進一步產生氧化作用使接枝到石墨層表面的磺酸基團分解,起到增強氧化和除雜的作用。
四是在石墨氧化剝離過程中,充分利用機械攪拌與超聲波疊加使用所產生的機械剪切效應、空化效應、化學強化效應以及熱效應作用,一方面強化石墨層間碳原子的氧化反應,從化學反應角度拉大石墨層間距,另一方面破壞原子間作用力并加速剝離碎片在溶液中的運動,從機械作用角度實現石墨的深度剝離與均勻分散。
五是在石墨氧化剝離反應完畢之后,繼續利用機械攪拌與超聲波疊加使用所產生的機械剪切效應、空化效應及熱效應作用進行深度剝離,進一步破壞原子間作用力以獲得尺寸更小、厚度更薄的單層石墨烯片,同時加速石墨碎片在介質中的運動,防止團聚,以獲得均勻分散的石墨烯分散液。
與現有技術相比,本發明的創新點在于:
1、本發明將傳統反應過程中石墨氧化階段采用的KMnO4氧化劑替換為臭氧(O3)強氧化劑,一方面充分利用強氧化性氣體的可流動性來強化傳質過程,加速石墨的插層氧化反應,有利于增大層間距;另一方面可大幅減少雜質離子的引入,克服傳統制備工藝中存在的金屬離子難清洗的問題,所得產品的純度高;
2、本發明充分發揮化學作用和機械作用的疊加累積效應,借助機械作用強化石墨碳原子的化學氧化作用,并在化學氧化的基礎上加速機械剪切剝離與分散傳質,二者相輔相成,互相促進,實現石墨的深度剝離與均勻分散,所得產品中1-2層石墨烯產率高達50%以上;
3、本發明實現各階段酸溶液的分步驟循環回用,無廢酸溶液外排,基本上規避了現有工藝所存在的廢酸排放與處理問題,大幅降低了該制備工藝的環境污染程度,同時還實現了硫酸資源的循環回用,降低了該制備工藝的經濟成本和環保成本。具體表現為:(1)本發明在完成石墨的氧化之后,采用離心的方式分離出大部分的硫酸溶液,向其中補加適量濃硫酸后可循環回用到石墨中溫氧化剝離階段;(2)在完成石墨深度剝離之后,采用離心方式分離出的上層稀酸溶液,可直接回用到石墨深度剝離階段;
4、由于本流程中不引入其他雜質離子,插層氧化過程中接枝的磺酸基團可在后期高溫氧化剝離階段進一步氧化水解脫除,所得產品純度高,無需多次洗滌即可達到純度大于99.9%的氧化石墨烯產品,避免了傳統制備工藝中多次洗滌也難以實現雜質深度脫除的問題。
總的來說,本發明提供了一條高效穩定、綠色環保的氧化石墨烯的制備工藝路線,克服了傳統酸氧化法存在的石墨烯活化難控制、產品純度不高等不足,制備得到的產品純度高(氧化石墨烯粉體的化學純度≥99.9%,比表面積為600~1500m2/g,1-2層氧化石墨烯產率≥50%)、安全性好、產品片層結構間距大、制備過程綠色環保。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖是本發明的一些實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。
圖1為本發明的氧化石墨烯的制備方法的工藝流程圖。
具體實施方式
為了便于理解本發明,下文將結合說明書附圖和較佳的實施例對本發明作更全面、細致地描述,但本發明的保護范圍并不限于以下具體的實施例。
除非另有定義,下文中所使用的所有專業術語與本領域技術人員通常理解的含義相同。本文中所使用的專業術語只是為了描述具體實施例的目的,并不是旨在限制本發明的保護范圍。
除非另有特別說明,本發明中用到的各種原材料、試劑、儀器和設備等均可通過市場購買得到或者可通過現有方法制備得到。
實施例1:
一種如圖1所示本發明的氧化石墨烯的制備方法,具體包括以下步驟:
(1)量取50ml 98%的濃硫酸于三口燒瓶中,然后置于冰水浴中降溫至0℃;
(2)開啟超聲波處理,精確稱取2g石墨粉(來自湖南郴州某石墨廠的天然鱗片石墨樣品,經選礦除雜處理后其化學純度為99.9%,粒度為40目~400目),超聲波處理的同時將石墨粉緩慢加入三口燒瓶中,在攪拌條件下同時輔以超聲處理,反應60min以進行低溫插層剝離;
(3)在上述步驟(2)中進行的低溫插層剝離完成后,持續超聲處理和攪拌不變,向所得的混合溶液中加入50ml 80%硫酸溶液,并持續向三口燒瓶中通入純度為90%的純氧源O3氣體作為氧化劑,氣體流速為2m3/h,然后緩慢水浴加熱使其逐步升溫到40℃環境進行恒溫攪拌反應60min,以實現中溫氧化剝離;
(4)在上述步驟(3)的中溫氧化剝離完成后,持續通入上述純氧源O3氣體作為氧化劑,然后開始向三口燒瓶加入30ml 30%的過氧化氫溶液,再次水浴加熱升溫到70℃高溫環境進行恒溫攪拌反應90min,以實現高溫氧化剝離;
(5)在上述步驟(4)的高溫氧化剝離完成后,停止通入含臭氧氣體,然后將所得的混合液進行離心處理,分離出上層清液(90ml)和下層懸浮液,分離出上層清液補充適量濃硫酸之后回用到上述步驟(3)的石墨中溫氧化剝離階段作為硫酸溶液補入,然后取45ml下層懸浮液于燒杯中,向其中加入200ml去離子水,持續超聲和攪拌處理使其深度反應60min,關閉攪拌和超聲波,得到亮黃色的氧化石墨烯分散液;
(6)將步驟(5)所得的氧化石墨烯分散液進行離心處理,得到的220ml上層清液可回用到上述步驟(5)中代替加入的去離子水以進行石墨的深度剝離,得到的下層沉淀加去離子水洗滌至中性后,經抽濾、真空干燥即得氧化石墨烯粉體。
經分析表征,本實施例所得的氧化石墨烯粉體產品的化學純度為99.95%,1-2層氧化石墨烯的產率為68%,產品的比表面積為924m2/g。
實施例2:
一種如圖1所示本發明的氧化石墨烯的制備方法,具體包括以下步驟:
(1)量取80ml 98%的濃硫酸于三口燒瓶中,然后置于冰水浴中降溫至-2℃;
(2)開啟超聲波處理,精確稱取4g石墨粉(來自湖南郴州某石墨廠的天然鱗片石墨樣品,經選礦除雜處理后其化學純度為99.92%,粒度為200目~1000目),超聲波處理的同時將石墨粉緩慢加入上述三口燒瓶中,在攪拌條件下同時輔以超聲處理,反應30min以進行低溫插層剝離;
(3)在上述步驟(2)中進行的低溫插層剝離完成后,持續超聲處理和攪拌不變,向所得的混合溶液中加入70ml 75%硫酸溶液,并持續向三口燒瓶中通入純度為60%的空氣源O3氣體作為氧化劑,氣體流速為5m3/h,然后緩慢水浴加熱使其逐步升溫到40℃中溫環境進行恒溫攪拌反應100min,以實現中溫氧化剝離;
(4)在上述步驟(3)的中溫氧化剝離完成后,持續通入上述空氣源O3氣體作為氧化劑,然后開始向三口燒瓶加入50ml 30%的過氧化氫溶液,再次水浴加熱升溫到80℃高溫環境進行恒溫攪拌反應60min,以實現高溫氧化剝離;
(5)在上述步驟(4)的高溫氧化剝離完成后,停止通入含臭氧氣體,然后將所得的混合液進行離心處理,分離出上層清液(150ml)和下層懸浮液,分離出上層清液補充適量濃硫酸之后回用到上述步驟(3)的石墨中溫氧化剝離階段作為硫酸溶液補入,然后取60ml下層懸浮液于燒杯中,向其中加入300ml去離子水,持續超聲和攪拌處理使其深度反應100min,關閉攪拌和超聲波,得到亮黃色的氧化石墨烯分散液;
(6)將步驟(5)所得的氧化石墨烯分散液進行離心處理,得到的320ml上層清液可回用到上述步驟(5)中代替加入的去離子水以進行石墨的深度剝離,得到的下層沉淀加去離子水洗滌至中性后,經抽濾、真空干燥即得氧化石墨烯粉體。
經分析表征,本實施例所得的氧化石墨烯粉體產品的化學純度為99.98%,1-2層氧化石墨烯的產率為71%,產品的比表面積為1258m2/g。
實施例3:
一種如圖1所示本發明的氧化石墨烯的制備方法,具體包括以下步驟:
(1)量取100ml 98%的濃硫酸于三口燒瓶中,然后置于冰水浴中降溫至2℃;
(2)開啟超聲波處理,精確稱取5g石墨粉(來自湖南長沙某石墨公司的天然鱗片石墨樣品,經選礦除雜處理后其化學純度為99.9%,粒度為200目~600目),超聲波處理的同時將石墨粉緩慢加入上述三口燒瓶中,在攪拌條件下同時輔以超聲處理,反應60min以進行低溫插層剝離;
(3)在上述步驟(2)中進行的低溫插層剝離完成后,持續超聲處理和攪拌不變,向所得的混合溶液中加入100ml 80%硫酸溶液,并持續向三口燒瓶中通入純度為70%的純氧源與空氣源O3混合氣體作為氧化劑,氣體流速為6m3/h,然后緩慢水浴加熱使其逐步升溫到45℃中溫環境進行恒溫攪拌反應100min,以實現中溫氧化剝離;
(4)在上述步驟(3)的中溫氧化剝離完成后,持續通入上述純氧源與空氣源O3混合氣體作為氧化劑,然后開始向三口燒瓶加入50ml 30%的過氧化氫溶液,再次水浴加熱升溫到80℃高溫環境進行恒溫攪拌反應120min,以實現高溫氧化剝離;
(5)在上述步驟(4)的高溫氧化剝離完成后,停止通入含臭氧氣體,然后將所得的混合液進行離心處理,分離出上層清液(180ml)和下層懸浮液,分離出上層清液補充適量濃硫酸之后回用到上述步驟(3)的石墨中溫氧化剝離階段作為硫酸溶液補入,然后取80ml下層懸浮液于燒杯中,向其中加入320ml去離子水,持續超聲和攪拌處理使其深度反應150min,關閉攪拌和超聲波,得到亮黃色的氧化石墨烯分散液;
(6)將步驟(5)所得的氧化石墨烯分散液進行離心處理,得到的350ml上層清液可回用到上述步驟(5)中代替加入的去離子水以進行石墨的深度剝離,得到的下層沉淀加去離子水洗滌至中性后,經抽濾、真空干燥即得氧化石墨烯粉體。
經分析表征,本實施例所得的氧化石墨烯粉體產品的化學純度為99.96%,1-2層氧化石墨烯的產率為54%,產品的比表面積為695m2/g。
實施例4:
一種如圖1所示本發明的氧化石墨烯的制備方法,具體包括以下步驟:
(1)量取80ml 98%的濃硫酸于三口燒瓶中,然后置于冰水浴中降溫至0℃;
(2)開啟超聲波處理,精確稱取5g石墨粉(來自湖南長沙某石墨公司的天然鱗片石墨樣品,經選礦除雜處理后其化學純度為99.9%,粒度為200目~600目),超聲波處理的同時將石墨粉緩慢加入上述三口燒瓶中,在攪拌條件下同時輔以超聲處理,反應60min進行低溫插層剝離;
(3)在上述步驟(2)中進行的低溫插層剝離完成后,持續超聲處理和攪拌不變,向所得的混合溶液中加入200ml 71%硫酸溶液(該硫酸溶液是由上述實施例3步驟(5)中離心分離出的180ml上層清液與20ml 98%濃硫酸混合配制而成),并持續向三口燒瓶中通入純度為60%的空氣源O3混合氣體作為氧化劑,氣體流速為5m3/h,然后緩慢水浴加熱使其逐步升溫到45℃中溫環境進行恒溫攪拌反應150min,以實現中溫氧化剝離;
(4)在上述步驟(3)的中溫氧化剝離完成后,持續通入上述空氣源O3混合氣體作為氧化劑,然后開始向三口燒瓶加入50ml 30%的過氧化氫溶液,再次水浴加熱升溫到70℃高溫環境進行恒溫攪拌反應150min,以實現高溫氧化剝離;
(5)在上述步驟(4)的高溫氧化剝離完成后,停止通入含臭氧氣體,然后將所得的混合液進行離心處理,分離出上層清液(250ml)和下層懸浮液,分離出上層清液補充適量濃硫酸之后可回用到上述步驟(3)的石墨中溫氧化剝離階段作為硫酸溶液補入,然后取90ml下層懸浮液于燒杯中,向其中加入350ml稀酸溶液(該稀酸溶液為上述實施例3步驟(6)分離出來的上層清液,其中H2SO4濃度為14.5%),持續超聲和攪拌處理使其深度反應240min,關閉攪拌和超聲波,得到亮黃色的氧化石墨烯分散液;
(6)將步驟(5)所得的氧化石墨烯分散液進行離心處理,得到的380ml上層清液可回用到上述步驟(5)中代替加入的稀酸溶液以進行石墨的深度剝離,得到的下層沉淀加去離子水洗滌至中性后,經抽濾、真空干燥即得氧化石墨烯粉體。
經分析表征,本實施例所得的氧化石墨烯粉體產品的化學純度為99.96%,1-2層氧化石墨烯的產率為51%,產品的比表面積為704m2/g。
- 還沒有人留言評論。精彩留言會獲得點贊!