制備硅納米材料的方法與流程
本發明涉及硅納米材料領域,具體涉及制備硅納米材料的方法。
背景技術:
基于鋰離子電池的需求不斷的提升,高性能的鋰離子電池電極材料的開發也受到了進一步的關注。硅納米材料由于具有高的理論儲鋰容量(-4200mAh/g,儲鋰容量是商用石墨負極材料的十倍多)和低的放電電位(<0.5V,Li/Li+),被認為是下一代高性能鋰離子電池比較關鍵的負極材料。目前,用于鋰離子電池的硅納米材料的制備得到了廣泛的研究。
已經提出了利用鎂熱還原法還原二氧化硅制備硅納米材料的方法(例如,Bao Z,Weatherspoon MR,Shian S等人,Nature,446:172~175(2007))。鎂熱還原法是使用適量的鎂,將二氧化硅還原為硅,同時生成氧化鎂。在此方法中,需要使用氫氟酸溶解殘余的二氧化硅。而眾所周知,氫氟酸具有腐蝕性并且對健康特別有害。
對于以更加簡便、環境友好、原料來源廣且成本低的方式獲得具備良好電極材料性能的硅納米材料的方法,仍存在需要。
技術實現要素:
為了解決上述問題,本發明提供了以下技術方案。
[1]一種制備硅納米材料的方法,其特征在于,所述方法包括以下步驟:
a)將含硅原料與過量的金屬研磨混合;
b)將a)中所得的混合物加熱至300至700℃進行反應,將所述含硅原料中的硅還原為金屬硅化物的形式;
c)將b)的反應產物在含氧氣體流下加熱至450至700℃進行反應,將其中的所述金屬硅化物氧化為單質硅核-二氧化硅殼的核-殼硅納米材料。
[2]根據[1]所述的方法,其特征在于,所述金屬選自由以下各項組成的組:鋰、鈉、鈣、鎂、鋅、鐵的單質或合金、以及它們的組合。
[3]根據[1]或[2]所述的方法,其特征在于,所述含硅原料中的硅與所述金屬的摩爾比為1∶3至1∶20。
[4]根據[1]至[3]中任一項所述的方法,其特征在于,在步驟b)中,反應溫度為450-650℃,反應時間為1-20h。
[5]根據[1]至[4]中任一項所述的方法,其特征在于,在步驟c)中,反應溫度為500-650℃,反應時間為1-20h。
[6]根據[1]至[5]中任一項所述的方法,其特征在于,在步驟c)中,所述含氧氣體是空氣或者是氧含量在1%~40%的氧氮、氧氬混合氣。
[7]根據[1]至[6]中任一項所述的方法,其特征在于,所述含硅原料為二氧化硅、硅酸、硅酸鹽、含硅礦物或者單質硅粉。
[8]根據[7]所述的方法,其特征在于,所述二氧化硅可以為處于以下形式的二氧化硅:硅砂、工業二氧化硅廢料、二氧化硅分子篩、白炭黑、玻璃纖維、微硅粉、以及它們的組合。
[9]根據[7]所述的方法,其特征在于,所述含硅礦物選自由以下各項組成的組:硅藻土、鈉長石、鉀長石、海泡石、活性白土、以及它們的組合。
[10]根據[7]所述的方法,其特征在于,所述含硅礦物預先經過預處理,所述預處理包括研磨和/或水洗和/或酸洗。
本發明的技術方案具有工藝操作簡單、成本低廉,易于擴大生產等優點。而且,本發明擴大了硅源的范圍,既可以直接使用工業二氧化硅廢料為原料,有效解決工業廢料的堆積和污染等問題;又可以直接使用含硅礦物為初始原料,提供了一種從礦物出發直接制備納米硅材料的有效路徑;而且無需使用四氯化硅等專門合成的原料作為硅源,降低了硅源的成本。合成過程中避免了使用氫氟酸、有毒有機溶劑等高污染物質,環境友好,易于工業生產。所合成的硅納米粉可以直接用于鋰離子電池負極材料中,具有電化學性能優良等特點,為實現高性能納米硅負極材料宏量制備提供了可能。
附圖說明
圖1是實施例1中過程產物的X射線衍射圖。由上到下依次為:工業二氧化硅原料的XRD圖;高溫釜內反應后的XRD圖;通氣煅燒后的XRD圖;清洗后的最終產物XRD圖。
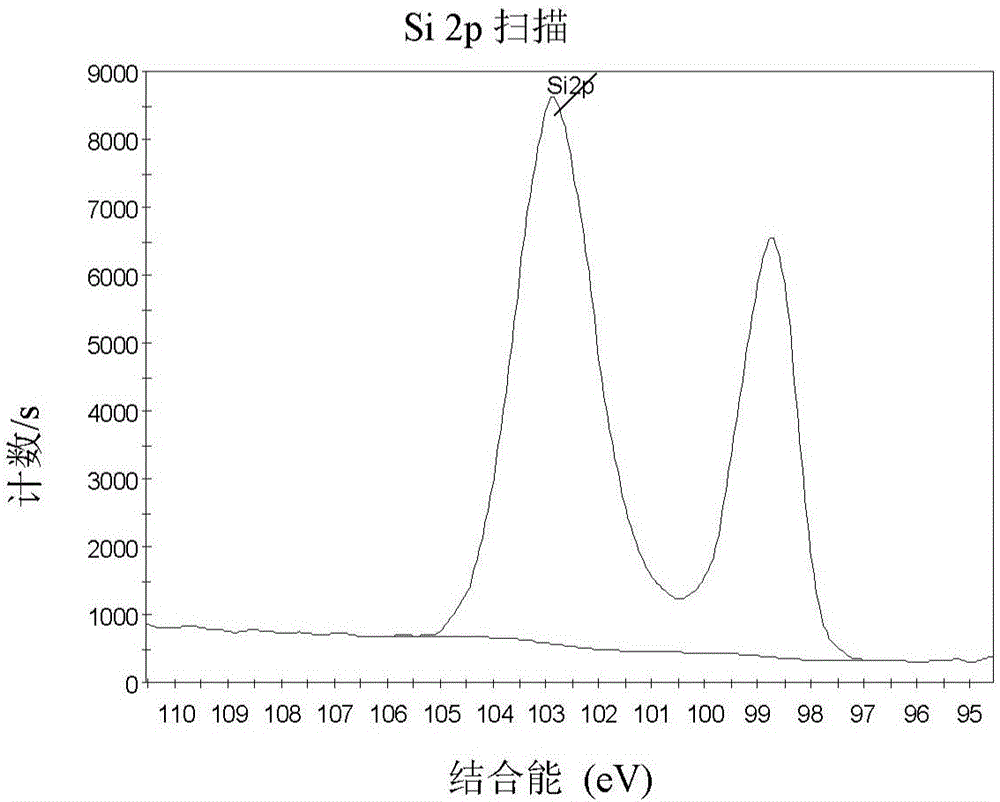
圖2是實施例1得到的最終產物X射線熒光光譜圖。
圖3是實施例1得到的最終產物掃描電鏡圖。
圖4是實施例1得到的最終產物透射電鏡圖。
圖5是實施例1得到的最終產物高分辨透射電鏡圖。
圖6是實施例2得到的產物X射線衍射圖。
圖7是實施例2得到的最終產物掃描電鏡和透射電鏡圖。
圖8是實施例3使用的5L不銹鋼反應釜示意圖和實物照片。
圖9是實施例3得到的產物X射線衍射圖。
圖10是實施例3得到的最終產物掃描電鏡圖。
圖11是實施例3得到的最終產物透射電鏡圖。
圖12是實施例3得到的最終產物高分辨透射電鏡圖。
圖13是實施例4得到的X射線衍射圖。
圖14是應用例1得到的硅納米粉體特征的充放電曲線圖。
圖15是應用例1得到的硅納米粉體的電化學循環穩定圖。
圖16是應用例2得到的硅納米粉體的電化學倍率性能圖,1C=3600mA/g。
圖17是應用例2得到的硅納米粉體的電化學循環穩定圖,1C=3600mA/g。
具體實施方式
本發明提供了一種制備硅納米材料的方法,其特征在于,所述方法包括以下步驟:a)將含硅原料與過量的金屬研磨混合;b)將a)中所得的混合物加熱至300至700℃進行反應,將所述含硅原料中的硅還原為金屬硅化物的形式;c)將b)的反應產物在含氧氣體流下加熱至450至700℃進行反應,將其中的所述金屬硅化物氧化為單質硅核-二氧化硅殼的核-殼硅納米材料。
在本發明的步驟a)中所稱的“過量的金屬”指的是金屬的量足夠多,使得在b)步驟反應后,仍有金屬剩余。例如,當含硅原料為二氧化硅時,在步驟a)中使用過量的金屬,如鎂,使得在b)步驟中的條件下反應后,二氧化硅中的硅與鎂形成硅化鎂,二氧化硅中的氧與鎂形成氧化鎂,并且還剩余未反應的鎂。這與現有技術如鎂熱反應是不同的。在典型的鎂熱反應中,鎂不是“過量”的,二氧化硅與鎂反應,直接生成單質硅,但不剩余未反應的鎂。
因此,本發明的方法中,含硅原料中的硅首先在步驟b)中被充分還原成金屬硅化物的形式,然后金屬硅化物在步驟c)中再被氧化成單質硅的形式。通過此先還原再氧化的過程,自然形成納米級的硅材料。
所述的研磨混合可以是采用研缽研磨或者采用球磨機磨成均勻粉末。對于粉末的粒度沒有特別的要求。雖然原料粉末不是納米粉末,但通過本發明的方法制得的硅粉末是納米粉末。
本發明的方法不在溶劑中進行。在步驟b)中,反應可以在密封的反應器內進行;當然,該反應也可以在惰性氣體保護的條件下進行。密封的反應器可以是密封的不銹鋼高溫釜。典型地,在本發明的方法中使用的高溫釜的體積為20mL-50L。
本發明的方法的步驟c)中最終得到的產物是單質硅核-二氧化硅殼的核-殼硅納米材料。在伴隨加熱通過含氧氣體的條件下,當將全部金屬硅化物氧化為單質硅后,在單質硅納米材料表面會形成二氧化硅膜。研究已經發現,在某些應用中,特別是在作為鋰離子電池負極的應用中,在硅粒子表面包覆一層二氧化硅是有利的。在這一點上,本發明的制備方法可以無需單獨的步驟便自然在硅納米粒子表面形成一層二氧化硅,是十分有利的。而且,采用本發明的方法,可以便捷地在步驟c)中通過調節含氧氣體、反應時間、溫度等具體條件,來影響二氧化硅殼的性質。
盡管二氧化硅殼層的存在在負極材料的應用中是有利的,但應理解,它與單質硅核相比在量上要少得多。二氧化硅殼的厚度通常為約幾nm,如小于5nm,可以為約2-3nm,而硅納米材料的粒徑通常為約幾十至幾百nm。
當然,根據實際需要,也可以對根據本發明的方法獲得的產品進行進一步加工。
本發明的方法中不需要使用在現有的納米硅材料制備中常用到的氫氟酸、有毒有機溶劑等高污染物質,環境友好,易于工業生產,并且所合成的硅納米粉的電化學性能優良,可以直接用于鋰離子電池負極材料中。
在本發明的方法中所述的金屬是能夠將硅還原為金屬硅化物的金屬。優選地,金屬選自由以下各項組成的組:鋰、鈉、鈣、鎂、鋅、鐵的單質或合金、以及它們的組合。上述金屬是常見的、具有足夠反應性的并且經濟上可行的金屬。
在本發明的方法中,金屬為過量即可。優選地,含硅原料中的硅與所述金屬的摩爾比為1∶3至1∶20。在此范圍內,既可以保證金屬滿足本發明方法的過量要求,又不至于金屬過量太多造成浪費。
在本發明的步驟b)中,反應時間通常為1-36h;優選的反應溫度為450-650℃;優選的反應時間為1-20h,如約10h。溫度過低,反應速度慢;溫度過高,能量消耗大。時間過短,反應不充分;時間過長,效率低下,產生時間和能量的浪費。
在本發明的步驟c)中,反應時間通常為1-72h;優選的反應溫度為500-650℃;優選反應時間為1-20h,更優選5-10h。溫度過低,反應速度慢;溫度過高,能量消耗大。時間過短,反應不充分;時間過長,效率低下,產生時間和能量的浪費。優選的反應時間也有利于形成對鋰離子電池負極性能有利的二氧化硅殼。
步驟c)中所述含氧氣體是指含有氧氣的氣體,例如純氧。優選地,所述含氧氣體可以是空氣或者是氧含量在1%~40%的氧氮、氧氬混合氣。如上所述,在步驟c)的含氧氣氛和溫度條件下,最終生成的硅納米材料表面有一層二氧化硅殼(膜)。當用于鋰電池負極中時,該二氧化硅膜可以提升電池的性能。
本發明的含硅原料包括常見的含有硅的材料。優選地,含硅原料為二氧化硅、硅酸、硅酸鹽、含硅礦物或者單質硅粉。
優選地,二氧化硅可以為處于以下形式的二氧化硅:硅砂、工業二氧化硅廢料、二氧化硅分子篩、白炭黑、玻璃纖維、微硅粉、以及它們的組合。直接使用工業二氧化硅廢料為原料尤其可以有效解決工業廢料的堆積和污染等問題,提供了工業二氧化硅廢料的有用的用途。
可以使用儲量豐富的含硅礦物為原料,直接制備納米硅粉,工藝操作簡單、成本低廉,易于擴大生產。含硅礦物優選選自由以下各項組成的組:硅藻土、鈉長石、鉀長石、海泡石、活性白土、以及它們的組合。
也可以直接使用單質硅粉作為含硅原料。本發明的方法提供了一種簡單高效的方式,從單質硅粉制備納米硅粉體材料。
本發明無需使用四氯化硅等專門合成的原料作為硅源,擴大了硅源的范圍,降低了硅源的成本。
在本發明的方法中,部分含硅礦物首先經過研磨、水洗和/或酸洗等預處理初步去除雜質。更具體地,所述預處理過程為如下:將含硅原礦用例如錘磨機機械地磨碎或碾碎成均勻粉末,然后與水按質量比1∶1~100,更優選為1∶4~50混合浸泡清洗足夠時間例如0.5~4h。然后,在上清液傾倒后,加入無機酸溶液例如0.5M的鹽酸溶液在常溫下攪拌1~20小時,優選5~10小時后,靜置1~50小時,優選2~24小時。最后經洗滌干燥,得到清洗后的含硅礦物。需說明的是,在本發明方法中,作為原料的含硅礦物也可以不經過預處理,尤其是以硅藻土、活性白土等作為原料時完全可以不需經過上述預處理過程。
本發明的步驟c)后的產物顯然還包括金屬氧化物等。但如本領域人員可以想到的,可以對本發明的產物進行后續的酸洗、分離、洗滌、干燥等步驟,分離出硅納米粒子粉末。例如,可以用稀鹽酸清洗步驟c)的產物,將其中的金屬氧化物(例如氧化鎂)等溶解,并且通過離心等方式分離固體硅納米材料,隨后水洗并干燥,來獲得硅納米粒子。
通過本發明的方法制備的硅納米材料可以用于硅納米材料的各種用途。特別地,本發明得到的硅納米粒子的電學性質優良,可以用作鋰離子電池的負極材料。
以下將通過具體實施例對本發明做進一步詳述,但應理解,這些實施例僅用于舉例說明的目的,而不用于限制本發明的范圍。
實施例1:以工業二氧化硅廢料為原料制備硅納米材料。
取5克工業二氧化硅(中國天信集團股份有限公司)與10克鎂粉研磨混合均勻后,放入50mL不銹鋼高溫釜內,密封并置于馬弗爐內500℃反應10h,然后自然冷卻至室溫,開釜后把反應產物倒入坩堝內并置于馬弗爐,在600℃下反應5h。所得產物經1M稀鹽酸清洗,水洗并采用高速離心機在4000轉/分鐘的轉速下離心5分鐘,倒去上清液并水洗離心,如此循環三次至上清液pH值為7,水洗后的樣品放置在真空干燥箱中80℃干燥過夜,即獲得2.2克硅納米粉,產率為94%。
采用X光粉末衍射儀(Philips X’Pert)對不同反應階段的產物進行X光衍射分析,圖1為該實施例所得過程產物的X射線衍射譜。其中由上到下依次為:(1)工業二氧化硅廢料的X光衍射譜圖,由圖可見該工業二氧化硅廢料為非晶相二氧化硅;(2)采用過量鎂粉在高溫釜內還原后所得到的產物的X光衍射譜圖,由圖可見該產物主要由硅化鎂、氧化鎂以及沒反應完的鎂組成;(3)在空氣中煅燒后的產物的X光衍射譜圖,由圖可見該產物主要由氧化鎂和硅組成,沒有了硅化鎂的峰,證明了該過程主要是硅化鎂在氧氣中氧化生成氧化鎂和單質硅;(4)清洗后的產物的X光衍射譜圖,由圖可見,X光衍射譜圖中2θ在10-80°范圍內有清晰可見的衍射峰,所有衍射峰均可指標為立方的Si(JPCDS 77-2111)。結合X射線熒光光譜(XPS,圖2)分析,顯示清洗后的產物為硅納米粉體,表面分布微量的無定型的二氧化硅。
最終產物的掃描電鏡圖(圖3)和透射電鏡圖(圖4)顯示該條件制備的硅粉為納米多孔結構,孔均勻分布在幾個納米范圍內。高分辨透射電鏡圖(圖5)顯示所獲得的材料表面包覆一層無定型的二氧化硅膜,厚度約3nm。
實施例2:以硅藻土原礦為原料制備硅納米材料。
如實施1所述的方案,取1克硅藻土(二氧化硅含量約為90%,臨江市亨泰助濾劑有限公司)與2克鎂粉研磨混合均勻后,放入20mL不銹鋼高溫釜內,密封并置于馬弗爐內500℃反應10h,然后自然冷卻至室溫,開釜后把反應產物倒入坩堝內并置于馬弗爐,在600℃下反應10h。所得產物經1M稀鹽酸清洗,水洗并采用高速離心機在4000轉/分鐘的轉速下離心5分鐘,倒去上清液并水洗離心,如此循環三次至上清液pH值為7,水洗后的樣品放置在真空干燥箱中80℃干燥過夜,即獲得>0.37克硅納米粉,產率為88%以上。
對所獲得的硅納米粉進行X光衍射(圖6)、掃描電鏡和透射電鏡(圖7)分析,獲得與實施例1類似的結果。
實施例3:如實施例1所述,所不同的是采用單質硅粉為硅源,制備硅納米粉體。
如實施1所述的方案,取11.2克工業硅粉(200目,硅含量為99%,國藥集團)與20.2克鎂粉研磨混合均勻后,放入50mL不銹鋼高溫釜內,密封并置于馬弗爐內500℃反應10h,然后自然冷卻至室溫,開釜后把反應產物倒入不銹鋼盤并置于5L反應釜內,在空氣氣流下600℃反應10h(反應釜如圖8所示,101:溫度傳感器)。所得產物經1M稀鹽酸清洗,水洗并采用高速離心機在4000轉/分鐘的轉速下離心5分鐘,倒去上清液并水洗離心,如此循環三次至上清液pH值為7,水洗后的樣品放置在真空干燥箱中80℃干燥過夜,即獲得>10克硅納米粉,產率為90%以上。
對所獲得的硅納米粉進行X光衍射(圖9)、掃描電鏡(圖10)和透射電鏡(圖11)分析,獲得與實施例1類似的結果。高分辨透射電鏡圖(圖12)顯示所獲得的材料表面包覆一層無定型的二氧化硅膜,厚度約2-3nm。
實施例4:如實施1所述的方案,所不同的是采用硅酸為硅源,制備硅納米粉體。
如實施1所述的方案,取1.04克硅酸(二氧化硅含量約為77%,國藥集團)與2.1克鎂粉研磨混合均勻后,放入20mL不銹鋼高溫釜內,密封并置于馬弗爐內500℃反應10h,然后自然冷卻至室溫,開釜后把反應產物倒入坩堝內并置于馬弗爐,在600℃下反應5h。所得產物經1M稀鹽酸清洗,水洗并采用高速離心機在4000轉/分鐘的轉速下離心5分鐘,倒去上清液并水洗離心,如此循環三次至上清液pH值為7,水洗后的樣品放置在真空干燥箱中80℃干燥過夜,即獲得0.35克硅納米粉,產率為94%。
對所獲得的硅納米粉進行X光衍射(圖13)分析,獲得與實施例1類似的結果。表明本發明所述方法可以進一步擴展到使用硅酸以及硅酸鹽作為硅源。
應用例1:將所獲得硅納米粉用作鋰離子電池負極材料。
將上述實施例1中獲得的硅納米粉與海藻酸鈉膠粘劑、導電炭黑以質量比6∶2∶2(60mg∶20mg∶20mg)加入并取適量的水放到球磨罐中進行混合,250轉/分鐘球磨2小時成均勻漿料,將其涂于銅箔集流體上,在80℃真空干燥12小時后壓成負極極片。將該負極極片作為測試電極,鋰片為對電極,聚烯烴多孔膜(Celgard2400)為隔膜,以1M LiPF6的碳酸乙烯酯(EC)和碳酸二甲酯(DMC)(體積比1∶1)及10%氟代碳酸乙烯酯(FEC)添加劑的混合溶液作為電解液(珠海賽維),組裝成CR2016扣式電池,該CR2016電池的組裝在氬氣氣氛的手套箱中完成。組裝得到的CR2016扣式電池在室溫下進行鋰離子電池儲鋰性能測試。充放電截止電壓為0.005-1.5V。圖14為該硅納米粉體特征的充放電曲線圖;圖15為該硅納米粉的電化學循環穩定性圖。從圖中可以看出,在3.6A/g的電流密度下,1000個循環圈內容量穩定保持在1000mAh/g以上。由此可知,利用本發明方法的實施例1制備的硅納米粉,能夠作為高性能的鋰離子電池負極材料。
應用例2:將實施例3所獲得硅納米粉用作鋰離子電池負極材料。
將上述實施例3中獲得的硅納米粉與海藻酸鈉膠粘劑、導電炭黑以質量比6∶2∶2(60mg∶20mg∶20mg)加入并取適量的水放到球磨罐中進行混合,250轉/分鐘球磨2小時成均勻漿料,將其涂于銅箔集流體上,在80℃真空干燥12小時后壓成負極極片。將該負極極片作為測試電極,鋰片為對電極,聚烯烴多孔膜(Celgard 2400)為隔膜,以1M LiPF6的碳酸乙烯酯(EC)和碳酸二甲酯(DMC)(體積比1∶1)及10%氟代碳酸乙烯酯(FEC)添加劑的混合溶液作為電解液(珠海賽維),組裝成CR2016扣式電池,該CR2016電池的組裝在氬氣氣氛的手套箱中完成。組裝得到的CR2016扣式電池在室溫下進行鋰離子電池儲鋰性能測試。充放電截止電壓為0.005-1.5V。圖16為該硅納米粉的倍率性能圖;圖17為該硅納米粉的電化學循環穩定性圖。從圖中可以看出,在36A/g的電流密度下,容量為1000mAh/g以上;在1.8A/g的電流密度下,400個循環圈內容量穩定保持在1200mAh/g以上。由此可知,利用本發明方法的實施例3制備的硅納米粉,能夠作為高性能的鋰離子電池負極材料。
實施例結果表明,本發明方法使用價格低廉的原料出發,實現了硅納米粉體的制備,并且產率高于90%。適當的放大試驗證明該方法具有易于擴大生產的可能。所獲得的硅納米粉體用于鋰離子電池負極材料時,顯示出遠高于石墨負極的儲鋰容量以較好的循環穩定性,可作為潛在的下一代高性能鋰離子電池負極材料。
- 還沒有人留言評論。精彩留言會獲得點贊!