一種從錳銀礦中回收錳和銀的方法與流程
本發明涉及濕法冶金
技術領域:
,更具體涉及一種從錳銀礦中回收錳和銀的方法。
背景技術:
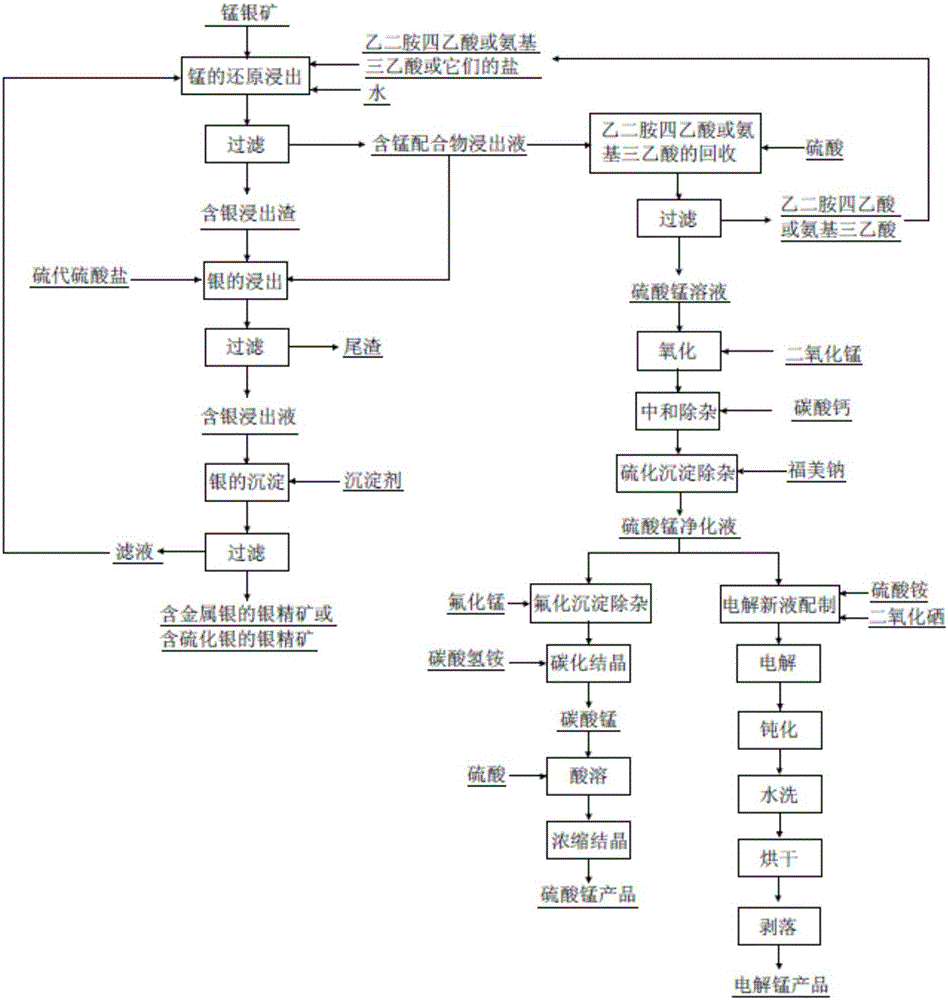
:含銀錳礦簡稱錳銀礦,錳品位一般為5%~35%,銀品位為10~104g/t,是重要的銀礦資源之一。近年來,我國的廣西、河北、江西、內蒙古、山西、湖南、云南、安徽、湖北、廣東、福建及北京等地相繼發現了大量的錳銀礦床,已探明的錳銀礦合計銀儲量已近萬噸。對錳銀礦的利用引起了研究者的重視,并且也取得了一定的進展。但由于錳銀礦的礦物性質比較復雜,尚未得到有效的開發和利用。錳銀礦所含錳按礦物類型可簡單劃分為氧化型、硫化型、碳酸鹽型(又稱菱型)及氫氧化型,其中,氧化型是目前探明的主要含錳類型,且主要在地表或淺層分布,是目前易經濟開采利用的主要類型。對氧化型錳銀礦所含錳礦物進一步鑒定,又分為軟錳礦、硬錳礦、褐錳礦及錳鉀礦等型,也伴生有少量的水錳礦、六方錳礦、錳鉛礦、菱錳礦、鐵菱錳礦及錳方解石等礦物,氧化型錳銀礦中的錳以MnO2為主要存在形式。錳銀礦所含銀礦物的形式及礦化形態、產出組合、結構、粒度及嵌布類型等與原礦成礦溶液的性質和氧化程度關系密切。銀的賦存形式主要有:以獨立銀礦物(自然銀、銀金礦、角銀礦及自輝銀礦等)形式存在;以類質同像形式分布在錳礦中;以微細粒礦物包體分布在錳礦或伴生鐵礦等集合體的微裂縫中;以離子吸附狀態存在等。錳和銀的這些賦存形式及分配比例,在不同產地的氧化型錳銀礦中有一定差異,但銀多以類質同像或獨立銀礦物分布為主。通常錳礦物在銀礦物表面形成了致密的包裹層,浸出劑難以擴散吸附于銀礦物表面進行反應。因此,采用直接氰化法、硫脲法或硫代硫酸鹽法等難以將銀直接溶出,必須破壞氧化錳礦物的晶體結構,才能進行銀的提取。在錳的處理方面,由于錳銀礦中的錳礦物常以高價態錳的氧化物的形式存在,難于直接進行酸浸,因此需要先進行還原處理,然后再進行酸浸。在銀的處理方面,主要以氰化浸銀法、硫脲浸銀法及硫代硫酸鹽浸銀法為主。中國專利文件CN1079258A公開了一種由錳銀礦直接生產硫酸錳和提取銀的方法。先用二氧化硫和硫酸還原浸出錳,采用氧化—中和法除雜,直接生產硫酸錳。然后氰化浸出提銀,再使用鋅粉置換,得到銀渣。中國專利文件CN1186866A公開了一種高錳硫鐵銀礦濕法提銀工藝。該工藝首先對浮選精礦進行氧化焙燒,再用硫酸將脫硫、脫砷后的精礦浸出,使用氯化鈉將銀以氯化銀的形式沉淀出來,最后將錳從沉銀后的浸出液中以碳酸錳的形式進行回收。中國專利文件CN1632143A公開了一種從錳銀精礦用氯化焙燒、氨浸出提取白銀和錳產品的方法。該方法首先將錳銀精礦進行氧化焙燒,然后進行氯化焙燒或直接氯化焙燒,然后對焙砂水洗去除可溶于水的金屬雜質,再將水洗后的焙砂與氨液混合攪拌浸出銀,銀氨溶液用水合肼還原,生產海綿銀。中國專利文件CN101831544A公開了一種錳銀礦錳、銀分離及其浸錳液的凈化處理方法。該方法首先進行植物副產秸稈、殼、渣預降解糖化工藝,得到的產物與錳銀原礦或富集后的混合精礦浸出錳。浸錳液經分離、中和、吸附處理得到可進一步凈化、結晶制備硫酸錳等的溶液。最后,將含銀浸錳渣浸出、還原回收銀,得到含銀粉。中國專利文件CN102703726A公開了一種通過磁選實現氧化型錳銀礦中錳和銀的富集,銀錳精礦粉壓制成銀錳精礦磚進行烘焙后,利用飽和氯化鈉溶液作為銀的浸出劑進行浸出,得到含銀浸出液和錳精礦磚的方法。中國專利文件CN104131182A公開了一種使用焦亞硫酸鈉在酸性介質中攪拌還原錳銀精礦,礦漿反應,還原反應完成后,過濾、固液分離,分別得到含錳溶液和含銀礦泥,后續浸錳液經深度凈化除雜制備純凈硫酸錳溶液,富銀渣使用氨性硫代硫酸鹽法提銀的方法。中國專利文件CN104561580A公開了一種從難選冶的氧化型錳銀礦中,使用硫酸亞鐵還原酸浸錳,氰化提銀的方法。另據文獻報道,黃鐵礦、閃鋅礦及砷金礦等硫化礦物,秸稈、淀粉及纖維素等生物質,以及過氧化氫、苯胺、苯酚、硫酸亞鐵、二氧化硫、亞硫酸鈉、焦亞硫酸鈉及煤粉等均可作為還原劑對錳銀礦中的氧化錳進行還原。在錳的還原酸浸過程中,可以輔以一定頻率和功率的超聲波或微波予以強化(袁明亮,邱冠周,王淀佐。細粒嵌布錳銀礦浸取中的超聲強化作用[J].過程工程學報,2002,2(1):21-25.)。在銀的浸出過程中,文獻報道了中溫氯化焙燒—氰化提銀法(蘇成德,趙禮兵,安雪梅.從難選錳銀礦尾礦中回收銀的研究[J].中國礦業,2008,17(1):89-92)及在CaCl2-HCl體系下浸取銀的方法(QinghuaTian,CuiyanJiao,XueyiGuo.Extractionofvaluablemetalsfrommanganese-silverore.Hydrometallurgy,2012,119-120:8-15.)。另外,亦有文獻報道了在硫酸體系中加入過氧化氫,實現同時對錳的還原浸出和對銀的氧化浸出的一步浸取法(JiangT,YangY,HuangZ,etal.Simultaneousleachingofmanganeseandsilverfrommanganese–silveroresatroomtemperature[J].Hydrometallurgy,2003,69(s1–3):177-186.)。上述文獻報道的方法,在技術上均具有一定的特點和優勢,但考慮到經濟成本和環境成本等問題,也均具有一定的限制。因此,對錳銀礦高效、低毒、低成本的開發與利用,仍需進一步的研究。技術實現要素:(一)要解決的技術問題本發明要解決的技術問題就是如何高效、低毒、低成本的從錳銀礦中回收錳和銀,而提供一種從錳銀礦中回收錳和銀的方法。(二)技術方案為了解決上述技術問題,本發明提供了一種從錳銀礦中回收錳和銀的方法,該方法包括以下步驟(所用原料均市購獲得):步驟一:錳的還原浸出:將乙二胺四乙酸或氨基三乙酸或它們的鹽和錳銀礦在水中攪拌發生還原浸出反應,反應完畢后過濾分離,得到含銀浸出渣和含錳配合物浸出液;步驟二:銀的催化浸出:將步驟一得到的含銀浸出渣加入到含硫代硫酸鹽的溶液中,再加入一部分含錳配合物浸出液進行銀的催化浸出,浸出完畢后過濾,得到含銀浸出液和尾渣;步驟三:銀的沉淀:將金屬錳粉、鋅粉、鐵粉、銅粉、鋁粉或連二亞硫酸鹽中的一種或幾種加入到含銀浸出液中得到含金屬銀的銀精礦;或將硫化鈉、硫氫化鈉、硫化銨、硫氫化銨、硫化鈣或硫化鋇中的一種或幾種加入到含銀浸出液中得到含硫化銀的銀精礦;步驟四:乙二胺四乙酸或氨基三乙酸的回收:將另一部分含錳配合物浸出液用硫酸酸化,沉淀、過濾,得到乙二胺四乙酸(EDTA)或氨基三乙酸(NTA)和硫酸錳溶液,將乙二胺四乙酸或氨基三乙酸循環使用;步驟五:硫酸錳溶液的除雜及錳產品的制備:將步驟四得到的硫酸錳溶液進行中和除雜及硫化沉淀除雜,得到硫酸錳凈化液,電解得到金屬錳;或將得到的硫酸錳凈化液進行氟化沉淀除雜、碳化結晶、酸溶及濃縮結晶,生產硫酸錳產品。優選地,在步驟一中,所述的錳的還原浸出反應的液固質量比為3~15∶1,乙二胺四乙酸或氨基三乙酸或他們的鹽和錳銀礦中錳的摩爾比為0.5~1.5∶1;所述的錳的還原浸出反應時間為0.5~5h,還原浸出溫度為20~95℃,還原浸出反應溶液pH值為4~9。優選地,在步驟二中,所述的銀的催化浸出的液固質量比為3~20∶1,含硫代硫酸鹽溶液的濃度為0.1~2mol/L,以錳摩爾濃度計錳配合物濃度為0.01~0.5mol/L;所述銀的催化浸出反應時間為0.5~5h,反應溫度為20~95℃,溶液pH值為4~9。優選地,在步驟三中,所述的銀的沉淀中錳粉、鋅粉、鐵粉、銅粉、鋁粉、連二亞硫酸鹽、硫化鈉、硫氫化鈉、硫化銨、硫氫化銨、硫化鈣或硫化鋇的用量為含銀浸出液中銀摩爾量的0.5~50倍;在20~95℃的溫度下攪拌沉淀銀,沉淀時間為0.5~5h。優選地,在步驟四中,所述的回收乙二胺四乙酸或氨基三乙酸中所加的硫酸與含錳配合物浸出液中錳的摩爾比為0.5~2∶1;所述的含錳配合物浸出液的酸化反應時間為0.5~5h,反應溫度為20~95℃。優選地,在步驟五中,所述的硫酸錳溶液的除雜及錳產品的制備的具體步驟為:(1)向硫酸錳溶液中加入二氧化錳,使二價鐵離子氧化為三價鐵離子,二氧化錳用量為二價鐵離子物質的量的0.5~1.5倍,反應溫度為30~80℃,反應時間為30~90min;(2)繼續加入碳酸鈣將浸出液的pH值調至4~6,除去硫酸錳溶液中的鐵、鋁離子;(3)使用福美鈉進行硫化沉淀除雜,福美鈉用量為0.02~0.10g/L,反應溫度為30~80℃,反應至溶液中的鋅離子不被檢出,除去硫酸錳溶液中的銀、銅、鎳、鉛及鋅重金屬離子,得到硫酸錳凈化液;(4)以硫酸錳凈化液為原料配制電解新液,使電解新液中含Mn:36~40g/L;(NH4)2SO4:110~130g/L;以Se計SeO2:0.03~0.04g/L;pH值=7~8.4,將該電解新液裝入電解槽,控制電解槽溫度35~40℃、陰極電流密度350~420A/m2、陽極電流密度600~700A/m2、槽電壓4.2~5.3V及電解周期2~24h,電解結束后,再經3%K2Cr2O7鈍化液鈍化、水洗、烘干及剝落步驟,得到電解錳產品;或者,在步驟五中,所述的硫酸錳溶液的除雜及錳產品的制備的具體步驟為:步驟(1)~(3)同上;(4)將硫酸錳凈化液進行氟化沉淀除雜,使用鈣、鎂離子摩爾總量1~1.5倍的氟化錳,在pH值6.5~7.5,溫度70~90℃下反應90~150min,除去硫酸錳凈化液中的鈣、鎂離子;(5)使用碳酸氫銨進行碳化結晶,緩慢滴加0.5~1.5mol/L碳酸氫銨溶液且用量為錳離子摩爾量的1~1.5倍,于30~50℃下攪拌反應30~90min,將氟化沉淀除雜后硫酸錳凈化液中的錳離子沉淀出來,與鈉離子和鉀離子分離,得到碳酸錳;(6)使用濃度為0.5~1.5mol/L硫酸溶液為底液,向其中緩慢加入制得的碳酸錳至溶液的pH值在5.5~6.5,然后在85~95℃的溫度下蒸發濃縮結晶,生產硫酸錳產品。優選地,所述的錳銀礦,即為含銀錳礦,錳品位為5%~35%,銀品位為10~104g/t;所述的錳銀礦還原浸出反應前破碎至20目以下。優選地,所述的乙二胺四乙酸或氨基三乙酸或它們的鹽包括乙二胺四乙酸、乙二胺四乙酸鈉鹽、乙二胺四乙酸銨鹽、乙二胺四乙酸鉀鹽、乙二胺四乙酸鈣鈉鹽、乙二胺四乙酸鎂鈉鹽、乙二胺四乙酸鋇鈉鹽、氨基三乙酸或氨基三乙酸鈉鹽。優選地,所述的連二亞硫酸鹽包括連二亞硫酸鈉、連二亞硫酸鉀、連二亞硫酸鈣或連二亞硫酸鋅中的一種或幾種。優選地,所述的硫代硫酸鹽包括硫代硫酸銨、硫代硫酸鈉、硫代硫酸鈣、硫代硫酸鉀、硫代硫酸鎂或硫代硫酸鋇中的一種或幾種。本發明實現的原理是:(1)錳的還原浸出:錳銀礦中的氧化錳與乙二胺四乙酸或氨基三乙酸或它們的鹽發生反應,被還原成可溶性的錳化合物,然后被乙二胺四乙酸或氨基三乙酸或它們的鹽絡合浸出。乙二胺四乙酸等還原浸出劑被氧化發生脫羧反應,生成乙二胺三乙酸、乙二胺二乙酸、乙二胺乙酸、氨基乙酸或繼續被氧化生成二氧化碳、水及甲醛等。以乙二胺四乙酸二鈉為例,發生的化學反應如式1~2所示。MnO2+EDTA2-→Mn2O3+Mn3O4+MnO+OD1+OD2+…+ODx(1)Mn2O3+Mn3O4+MnO+EDTA2→Mn(Ⅲ)(EDTA)-+Mn(Ⅱ)(EDTA)2-(2)式1中的OD1、OD2、…、ODx代表乙二胺四乙酸的氧化產物。(2)銀的催化浸出:乙二胺四乙酸錳或氨基三乙酸錳可作為催化劑用于銀的硫代硫酸鹽浸出,以乙二胺四乙酸錳為例,發生的化學反應如式3~6所示。4Ag+8S2O32-+2H2O+O2=4[Ag(S2O3)2]3-+4OH-(3)Ag+Mn(Ⅲ)(EDTA)-+2S2O32-=[Ag(S2O3)2]3-+Mn(Ⅱ)(EDTA)2-(4)AgCl+2S2O32-=[Ag(S2O3)2]3-+Cl-(5)Ag2S+Mn(Ⅱ)(EDTA)2-+4S2O32-=MnS+2[Ag(S2O3)2]3-+EDTA4-(6)(3)銀的沉淀反應:以錳粉、鋅粉、鐵粉、銅粉、鋁粉或連二亞硫酸鹽對含銀浸出液中的銀進行還原,得到含金屬銀的銀精礦;或以硫化鈉、硫氫化鈉、硫化銨、硫氫化銨、硫化鈣或硫化鋇對含銀浸出液中的銀進行沉淀,得到含硫化銀的銀精礦。以錳粉、鋅粉、鐵粉、連二亞硫酸鈉、硫化鈉及硫化鈣為例,發生的化學反應如式7~13所示。2[Ag(S2O3)2]3-+Mn=2Ag+Mn2++4S2O32-(7)2[Ag(S2O3)2]3-+Zn=2Ag+Zn2++4S2O32-(8)3[Ag(S2O3)2]3-+Fe=3Ag+Fe3++6S2O32-(9)2[Ag(S2O3)2]3-+Fe=2Ag+Fe2++4S2O32-(10)2[Ag(S2O3)2]3-+S2O42-+4OH-=2Ag+4S2O32-+2SO32-+2H2O(11)2[Ag(S2O3)2]3-+Na2S=Ag2S+2Na++4S2O32-(12)2[Ag(S2O3)2]3-+CaS=Ag2S+Ca2++4S2O32-(13)(4)乙二胺四乙酸或氨基三乙酸的回收:乙二胺四乙酸錳或氨基三乙酸錳可用硫酸酸化,沉淀、過濾,得到乙二胺四乙酸或氨基三乙酸和硫酸錳溶液,以乙二胺四乙酸錳為例,發生的化學反應如式14所示。Mn(Ⅱ)(EDTA)2-+2Na++2H2SO4=MnSO4+Na2SO4+EDTA·4H(14)(5)硫酸錳溶液的除雜及錳產品的制備:去除硫酸錳溶液中的雜質金屬離子及錳產品制備的代表性化學反應如式15~20。MnO2+2Fe2++4H+=2Fe3++Mn2++2H2O(15)Fe3++3OH-=Fe(OH)3(16)Zn2++S2-=ZnS(17)Mn2++2e=Mn(18)Ca2++2F-=CaF2(19)MnSO4+2NH4HCO3=MnCO3+(NH4)2SO4+CO2(g)+H2O(20)(三)有益效果(1)本發明使用乙二胺四乙酸或氨基三乙酸或它們的鹽同時作為還原劑和浸出劑,將錳銀礦中氧化錳一步還原浸出,且還原浸出錳的過程是在弱酸或中性或弱堿環境中進行的,有利于銀的氰化浸出或硫代硫酸鹽浸出。(2)錳銀礦中氧化錳還原浸出過程中所產生的含錳配合物可作為催化劑,用于銀的硫代硫酸鹽浸出,可替代傳統的銅氨催化體系且可以減小硫代硫酸鹽的消耗量,從而減少生產所用試劑種類及消耗,降低生產成本。(3)乙二胺四乙酸或氨基三乙酸或它們的鹽可通過酸化過程從還原浸出步驟中所產生的含錳配合物浸出液中沉淀回收,循環使用,從而降低試劑消耗及生產成本。(4)本發明配制的錳電解新液中殘留了少量的乙二胺四乙酸鹽,這些乙二胺四乙酸鹽在電解錳過程中,可以減少或防止陽極泥的產生,對電解過程有益。(5)本發明將錳的浸出反應和銀的浸出反應分步進行,方便分別對兩種浸出液進行處理,實現了錳和銀的高效浸出,錳和銀的浸出率分別可達97%和90%以上。(6)硫代硫酸鹽法作為高效低毒的金、銀浸出劑,具有廣闊的發展前景。附圖說明為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的一些實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。圖1是本發明從錳銀礦中回收錳和銀的工藝流程示意圖;圖2是本發明實施例3中的含金屬銀的銀精礦的X-射線衍射光譜圖;圖3是本發明實施例4中的電解錳產品的X-射線衍射光譜圖;圖4是本發明實施例5中的硫酸錳產品的X-射線衍射光譜圖。具體實施方式下面結合附圖和實施例對本發明的實施方式作進一步詳細描述。以下實施例用于說明本發明,但不能用來限制本發明的范圍。實施例中所有百分數除另有規定外均指質量百分數。實施例1國內某錳銀礦原礦,含錳17.61%、銀384g/t,將該礦破碎、磨礦至90%過120目。將200g該錳銀礦粉與187g乙二胺四乙酸在水中攪拌發生錳的還原浸出反應,液固質量比為5∶1,在70℃下反應1h,錳的浸出率為97.5%。還原浸出錳后經過濾得到含銀浸出渣及乙二胺四乙酸錳浸出液。向含銀浸出渣中加入適量水和一部分乙二胺四乙酸錳浸出液,以錳摩爾濃度計使得乙二胺四乙酸錳濃度達到0.05mol/L,再添加適量硫代硫酸銨,使得硫代硫酸鹽濃度達到0.3mol/L,液固質量比控制在5∶1,于40℃下攪拌3h,銀的浸出率為91.64%。測得浸銀反應結束后浸出液中硫代硫酸鹽濃度為0.22mol/L。最后向含銀浸出液中加入0.17g錳粉置換沉淀銀,在60℃下攪拌3h,銀的沉淀率為98.94%。或最后向含銀浸出液中加入0.15g硫化鈉硫化沉淀銀,在80℃下攪拌2h,銀的沉淀率為95.01%。本實施例得到了較高的錳浸出率及銀的浸出率和沉淀率。實施例2錳銀礦及操作步驟與實施例1相同,改變乙二胺四乙酸錳的濃度,其他浸出條件與實施例1相同,考察乙二胺四乙酸錳濃度對銀的浸出率的影響,結果如表1所示。結果表明,沒有添加乙二胺四乙酸錳溶液時,銀的浸出率僅有39.61%,而添加乙二胺四乙酸錳溶液時,銀的浸出率均在89%以上,說明乙二胺四乙酸錳對銀的浸出具有非常有益的效果。表1:乙二胺四乙酸錳濃度對銀浸出率的影響乙二胺四乙酸錳濃度(mol/L)銀的浸出率(%)039.610.0189.890.0591.640.1091.280.5091.18實施例3國內某錳銀礦精礦,含錳27.02%、銀451.5g/t,將該礦磨至90%過120目。將200g該錳銀礦粉與512g乙二胺四乙酸二鈉在水中攪拌發生還原浸出反應,液固質量比為3∶1,在95℃下反應1h,錳的浸出率為97%,乙二胺四乙酸錳浸出液中錳的濃度為87.36g/L,各雜質組分含量見表2。還原浸出錳后經過濾得到含銀浸出渣及乙二胺四乙酸錳浸出液。向含銀浸出渣中加入適量水和一部分乙二胺四乙酸錳浸出液,以錳摩爾濃度計使得乙二胺四乙酸錳濃度達到0.05mol/L,再添加適量硫代硫酸銨,使得硫代硫酸鹽濃度達到0.3mol/L,液固質量比控制在5∶1,于95℃下攪拌3h,銀的浸出率為90.03%,含銀浸出液中銀的濃度為77.15mg/L。測得浸銀反應結束后浸出液中硫代硫酸鹽濃度為0.26mol/L。向含銀浸出液中加入10g連二亞硫酸鈉還原沉淀銀,控制溶液pH值在6.4至6.7之間,并在40℃下攪拌1h,銀的沉淀率為96.05%。對所得的銀精礦進行檢測,X-射線衍射光譜分析結果如圖2所示,X-射線熒光光譜分析結果見表3;使用鉛試金富集硫氰酸鉀滴定法對產品中銀的含量進行測定,結果為36.50%。本實施例得到了一種高銀品位的銀精礦產品,其中銀主要以金屬銀的形式存在。表2:乙二胺四乙酸錳浸出液中各雜質組分含量元素FeAlCuNiCoPbZn濃度g/L1.980.70NDNDND0.671.11元素AgSrAsCaMgNaK濃度g/L0.0080.05ND1.370.0182.902.07ND:未檢出(檢出限:1μg/L)表3銀精礦的X-射線熒光光譜分析結果元素含量,%O14.8Si0.028S18.13Ca10.85Mn21.28Fe0.0428Se0.007Sr0.0506Ag34.82實施例4對實施例3中另一部分乙二胺四乙酸錳浸出液進行硫酸酸化回收乙二胺四乙酸,將114g濃硫酸(質量濃度為98%)與乙二胺四乙酸錳浸出液混合攪拌,酸化反應時間為2h,反應溫度為95℃,然后冷卻至室溫,測得乙二胺四乙酸回收率為70%。對酸化后所得的硫酸錳溶液進行除雜以生產電解錳產品。過程包括:(1)向硫酸錳溶液中加入0.5g二氧化錳,使二價鐵離子氧化為三價鐵離子,反應溫度為60℃,反應時間為60min;(2)使用碳酸鈣將浸出液pH值調至5.0左右,除去硫酸錳溶液中的鐵、鋁離子,中和除雜后溶液中各雜質組分含量如表4所示;(3)再向該硫酸錳溶液中加入0.04g福美鈉進行硫化沉淀除雜,反應溫度為70℃,反應至溶液中的鋅離子不被檢出,除去硫酸錳溶液中的銀、鉛、鋅等重金屬離子,得到硫酸錳凈化液,硫化沉淀除雜后溶液中各雜質組分含量如表5所示;(4)以步驟(3)中得到的硫酸錳凈化液為原料配制電解新液,使電解新液中含Mn:38g/L;(NH4)2SO4:120g/L;SeO2(以Se計):0.04g/L;pH值=7,將該電解新液裝入電解槽,控制電解槽溫度40℃、電解電流0.45A、電壓4.5V及電解周期2h,電解結束后,再經鈍化(3%K2Cr2O7鈍化液)、水洗、烘干及剝落等步驟,得到電解錳產品。對所得的電解錳產品進行檢測,X-射線衍射光譜分析結果如圖3所示,根據標準YB/T051-2003中提供的檢測方法,對電解錳產品各化學成分進行測定,其結果見表6,結果表明該電解錳產品能夠達到YB/T051-2003中牌號DJMnB的要求。本實施例通過硫酸酸化的方法回收了部分錳的還原浸出劑,且使用回收還原浸出劑后的溶液,通過除雜及電解等步驟生產出了符合要求的電解錳產品。表4:中和除雜后各雜質組分含量元素FeAlCuNiCoPbZn濃度g/LND0.19NDNDND0.310.28元素AgSrAsCaMgNaK濃度g/L0.0040.04ND0.870.0180.32.1ND:未檢出(檢出限:1μg/L)表5:硫化沉淀除雜后各雜質組分含量元素FeAlCuNiCoPbZn濃度g/LND0.18NDNDNDNDND元素AgSrAsCaMgNaK濃度g/LND0.04ND0.820.00981.12.04ND:未檢出(檢出限:1μg/L)表6:電解錳產品各化學成分測定結果元素MnSCSiPFeSe含量%99.900.040.0150.003NDND0.0008實施例5以實施例4步驟(3)中得到的硫酸錳凈化液為原料生產硫酸錳產品。過程包括:(1)將1.2g氟化錳加入到該硫酸錳凈化液中進行氟化沉淀除雜,在pH值為7.0,于90℃下反應120min,除去硫酸錳凈化液中的鈣、鎂離子,氟化沉淀除雜后溶液中各雜質組分含量如表7所示;(2)使用碳酸氫銨進行碳化結晶,緩慢滴加1mol/L碳酸氫銨溶液,共2L,于40℃下攪拌反應60min,將錳離子沉淀出來,與鈉離子和鉀離子分離,得到碳酸錳;(3)使用濃度為1.0mol/L硫酸溶液為底液,向其中緩慢加入制得的碳酸錳至溶液的pH值為6.0,然后在90℃左右的溫度下蒸發濃縮結晶,生產硫酸錳產品。對所得的硫酸錳產品進行檢測,X-射線衍射光譜分析結果如圖4所示,利用硫酸亞鐵銨滴定法測定產品中錳含量為32.0%滿足HG2936-1999飼料級硫酸錳大于31.8%的要求,另外,根據HG2936-1999提供的檢測方法,測定雜質離子含量、水不溶物含量及細度等指標均達到飼料級硫酸錳的標準。本實施例通過對回收還原浸出劑后的溶液進一步的除雜,以及蒸發結晶等步驟生產出了符合要求的硫酸錳產品。表7:氟化沉淀除雜后各雜質組分含量元素FeAlCuNiCoPbZn濃度g/LND0.05NDNDNDNDND元素AgSrAsCaMgNaK濃度g/LNDNDND0.0240.00379.971.99ND:未檢出(檢出限:1μg/L)對比例1錳銀礦與實施例1相同。錳的還原浸出方法與實施例1相同。向含銀浸出渣中加入NaCN溶液,控制NaCN的質量濃度為0.15%,液固質量比為3∶1,使用CaO作為保護堿,控制溶液pH值為10,于25℃下攪拌12h,銀浸出率為76.51%。該法銀的浸出率比硫代硫酸鹽催化浸銀法低,浸出所需時間長,而且氰化物浸出劑毒性大。對比例2錳銀礦與實施例1相同。將乙二胺四乙酸視為氧化錳的還原劑使用,加入量僅為實施例1中的30%。將還原渣與硫酸濃度為1.5mol/L、硫脲質量濃度為15g/L的溶液混合,于50℃下攪拌3h,控制反應的液固質量比為5∶1,得到含錳和銀的浸出液,錳的浸出率為98.16%,銀的浸出率為70.1%。乙二胺四乙酸可僅作為氧化錳的還原劑使用;對于該類錳銀礦,酸性硫脲浸銀體系效率不及本發明的硫代硫酸鹽催化浸銀體系。對比例3錳銀礦與實施例1相同。首先將Na2SO3和濃H2SO4(質量濃度為98%)與錳銀礦按質量比分別為0.92∶1及0.89∶1在水中攪拌發生還原浸出反應,控制該反應液固質量比為5∶1,在30℃下反應15min,錳浸出率為98.82%。過濾,洗滌,得到含銀浸出渣,然后向該含銀浸出渣中加入濃鹽酸(質量濃度為37%)和CaCl2,控制濃鹽酸與含銀浸出渣的質量比為0.66∶1,Cl-的濃度為300g/L,液固質量比為5∶1,并在85℃下攪拌4h,銀的浸出率為54.89%。該法浸錳效果較好,但銀的浸出效果不及本發明所提供的方法。以上實施方式僅用于說明本發明,而非對本發明的限制。盡管參照實施例對本發明進行了詳細說明,本領域的普通技術人員應當理解,對本發明的技術方案進行各種組合、修改或者等同替換,都不脫離本發明技術方案的精神和范圍,均應涵蓋在本發明的權利要求范圍當中。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1