一種碳點/金復合納米粒子的制備方法及用途與流程
本發明涉及碳與貴金屬的復合納米材料的制備及應用,特別涉及一種高量子產率小尺寸碳點/金(Cdot-Au)復合納米粒子的制備方法及其用途。
背景技術:
復合納米材料將不同功能的納米材料加以整合,可構建出具有多重功能的納米粒子,實現單一納米粒子無法完成的應用目標,是納米材料研制的重要方向之一。一方面,碳點是近年發展起來的一類新型碳納米材料,因具有熒光強度高、耐光漂白、無光閃爍、生物相容性好、易于修飾改性等諸多優點而備受關注。另一方面,金屬納米粒子具有特殊的光電性能,如光催化、光熱效應、表面等離子共振等,通過改變粒徑、形貌或化學環境,可調控金屬納米粒子的物理化學性能。為結合兩者的優勢,據報道,自2004年Xu等發現碳點以來,科研工作者設計了多種基于碳點的金屬復合納米材料,并探索了其在各個領域的應用,如胡勝亮等制備了碳/銀復合納米粒子并考察了其對亞甲基藍的可見光催化性能,康振輝等合成了碳/金復合納米材料并探討了對環己烷的光催化性能,張友玉等構建了碳/金復合納米粒子并應用于檢測氨基酸。上述工作表明,通過簡單混合的方法,調控實驗條件能夠獲得碳/金復合納米粒子。然而,所制備的復合納米粒子的量子產率遠低于碳點本身。此外,由于還原劑和穩定劑是不同物質,而碳點與金顆粒之間的作用力弱,異相成核作用有限,均難以抑制高表面能的金納米團簇向金納米顆粒的轉變,導致復合納米粒子粒徑大且分散。
技術實現要素:
本發明的目的在于克服上述現有技術存在的不足,提供一種高量子產率小尺寸碳點/金復合納米粒子的制備方法及其用途。本發明通過將強穩定和還原作用的官能團如巰基和氨基等同時引入小尺寸的碳點,并以碳點為異相成核中心,原位還原和穩定金顆粒,構建出高量子產率小尺寸的碳點/金復合納米粒子(Cdot-Au)。
本發明的目的是通過以下技術方案來實現的:
本發明涉及一種碳點/金復合納米粒子(Cdot-Au)的制備方法,所述方法包括如下步驟:
A、以檸檬酸和半胱氨酸為原料,制備氮硫共摻雜的碳點,從而將具有還原和穩定作用的官能團同時引入碳點;所述官能團包括羥基、羧基、氨基、巰基等;
B、將氯金酸的水溶液加入至所述碳點的水溶液中,以碳點為金納米顆粒異相成核中心、還原劑和穩定劑,發生原位還原反應,制得Cdot-Au粗產物;
C、所得粗產物經離心和透析純化后得到碳點/金復合納米粒子水溶液,烘干即得所述碳點/金復合納米粒子。
與現有的制備方法相比,上述制備方法中,由于碳點同時起到了成核、還原和穩定的作用,避免加入任何表面活性劑或吸附劑等,制備具有量子產率高、粒徑小、生物毒性小和耐光漂白等特點的復合納米粒子。同時制備方法也大大簡化。
作為優選方案,所述步驟A中,所述的氮硫共摻雜碳點量子產率大于20%,粒徑小于等于3nm。更優選量子產率大于50%。更優選粒徑為1~3nm。
所述步驟B中,所述氮硫共摻雜碳點的異相成核、還原和穩定等協同作用,制備可控的碳點/金復合納米粒子。
作為優選方案,所述步驟A中,氮硫共摻雜的碳點制備包括:
將0.5~3.5g檸檬酸與0.5~2g L-半胱氨酸加入10mL水中,攪拌至均勻混合,得混合溶液;
將所述混合溶液轉移至水熱反應釜中,于180~200℃馬弗爐中反應2~8h,冷卻至室溫;經離心、透析、過濾、干燥得純化后的碳點。
需要說明的是,本發明的碳點制備并不限于以上的水熱合成法;任何能制備出小粒徑(粒徑小于3nm)、引入了具有還原和穩定作用的官能團(羥基、羧基、氨基和巰基等)的碳點的方法都適用于本發明。正是由于小粒徑、以及引入了具有還原和穩定作用的官能團這一特性,本發明的碳點在本發明的Cdot-Au的制備中(步驟B)能夠同時起到成核、還原和穩定的作用。
作為優選方案,所述氮硫共摻雜的碳點制備中的離心、透析具體為:以3000~5000r/min離心10min,取上清液透析48h,采用的透析袋的截留分子量為3000Da。此時,制備得到的多數碳點粒徑為1~2.5nm,粒徑分布相當均勻,且呈單分散狀態。
作為優選方案,所述檸檬酸為無水或含結晶水的檸檬酸。
作為優選方案,步驟B中,氯金酸的水溶液中氯金酸的質量濃度為10~100μg/mL;所述氯金酸與碳點的質量比為1∶(0.2~4)。
作為優選方案,所述氯金酸與碳點的質量比為1∶(3~4)。
作為優選方案,步驟B中,所述原位還原反應是在N2保護條件下,在10~100℃的溫度條件下完成。溫度小于10℃反應進行過于緩慢,溫度大于100℃溶劑揮發嚴重且需運用 特殊設備。
作為優選方案,所述原位還原反應的時間為1~8h,反應時間過短還原反應不能得以完全進行,8h后反應已經完成,延長時間對產物無任何助益。
作為優選方案,步驟C中,所述碳點/金復合納米粒子中金元素的質量含量為3%~15%,碳點的質量含量為97%~85%。
本發明還涉及一種前述的制備方法制得的碳點/金復合納米粒子在細胞標記中的用途。
作為優選方案,所述碳點/金復合納米粒子與細胞共孵育,應用于細胞成像。
作為優選方案,所述細胞成像為癌細胞成像。還可應用于其他細胞系的成像。如引入合適的靶向修飾,則能實現亞細胞結構、細胞器或生物大分子的成像。
在本發明中,Cdot-Au粒徑為1~3nm。
在本發明中,Cdot-Au在340nm左右有一個寬的紫外吸收峰,對應熒光發射峰在400~500nm之間,當激發波長在300~400nm范圍內改變時,發射峰最強位置在425nm處保持不變。
在本發明中,Cdot-Au在水溶液中的量子產率為30%~60%,熒光壽命為11.8ns。
本發明以檸檬酸和半胱氨酸為原料,采取水熱法合成氮硫共摻雜碳點;以該碳點為金納米顆粒異相成核中心、還原劑和穩定劑,無需添加任何表面活性劑或吸附劑等助劑,通過一步還原氯金酸制備碳點/金復合納米粒子。與現有技術相比,本發明具有如下有益效果:
1、本發明利用高量子產率的碳點為異相成核中心、還原劑和穩定劑,在10~100℃條件下經原位還原氯金酸制備Cdot-Au;制備工藝簡便可行、反應條件溫和且綠色環保。所制備的碳點/金復合納米粒子具有量子產率高、尺寸小、生物毒性小和耐光漂白等特點,有望應用于細胞成像或催化反應研究等領域。
2、本發明制備的Cdot-Au具有量子產率高、尺寸小、生物毒性小和耐光漂白等特點,是已報道量子產率最高的碳/金復合納米材料;
3、本發明的Cdot-Au細胞毒性小,濃度達500μg/mL時,細胞存活率仍在70%以上,適合應用于生物醫藥領域。
附圖說明
通過閱讀參照以下附圖對非限制性實施例所作的詳細描述,本發明的其它特征、目的和優點將會變得更明顯:
圖1為本發明所述Cdot-Au和原料碳點的透射電鏡TEM圖,其中,圖a為原料碳點的TEM圖,圖b,c,d,e,f分別為原料碳點與氯金酸的質量比為0.2∶1,0.4∶1,2∶1,3∶1,4∶1 時所制備的Cdot-Au的TEM圖(對應實施例1和2的產物);
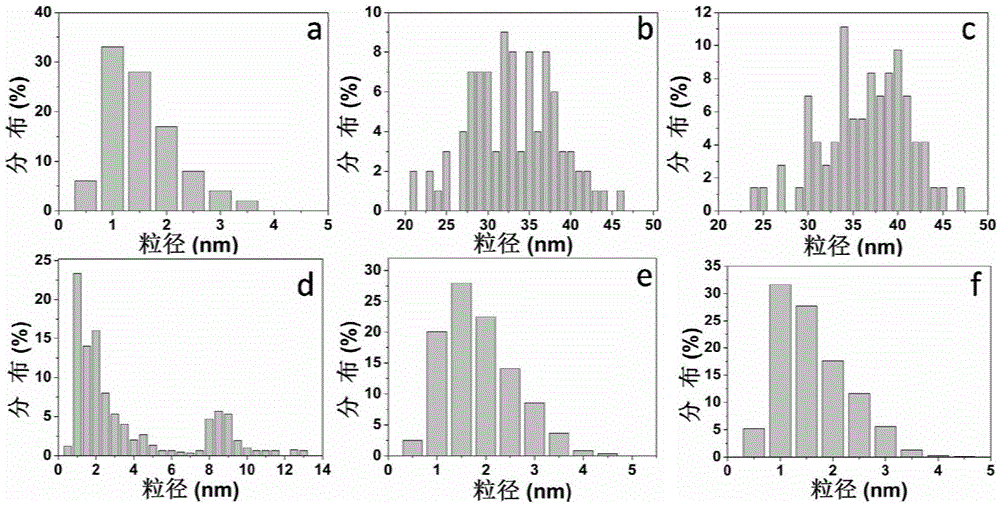
圖2為本發明所述Cdot-Au和原料碳點的粒徑分布圖,其中圖a為原料碳點的TEM圖,圖b,c,d,e,f分別為原料碳點與氯金酸的質量比為0.2∶1,0.4∶1,2∶1,3∶1,4∶1時所制備的Cdot-Au的粒徑分布圖(對應實施例1和2的產物);
圖3為本發明所述原料碳點的紫外可見吸收光譜與熒光光譜圖(對應實施例1的產物),其中,圖a為原料碳點的紫外吸收和熒光光譜圖,圖b是原料碳點用不同波長激發時的熒光發射譜圖;
圖4為本發明所述Cdot-Au與原料碳點的X射線衍射圖,圖中,a代表碳點的XRD分析圖,b代表Cdot-Au的XRD分析圖(對應實施例1和2的產物);
圖5為本發明所述Cdot-Au與原料碳點的XPS分析圖(對應實施例1和2的產物);其中,圖a為寬譜圖,圖b為C 1s高分辨譜圖及擬合曲線;
圖6為本發明所述Cdot-Au與原料碳點的熱重分析圖,圖中a表示Cdot-Au的TGA分析曲線,b表示原料碳點的TGA分析曲線(對應實施例2的產物);
圖7為本發明所述Cdot-Au與原料碳點的紅外光譜圖,圖中,a為原料碳點的紅外光譜圖,b為Cdot-Au的紅外光譜圖(對應實施例2的產物);
圖8為本發明所述Cdot-Au與原料碳點的紫外可見吸收和熒光譜圖,圖a中1表示Cdot-Au的紫外可見吸收光譜,2表示原料碳點的紫外可見吸收光譜圖,圖b表示Cdot-Au的紫外可見吸收和熒光發射光譜,圖中Cdot表示碳點,Cdot-Au表示Cdot-Au(對應實施例2的產物);
圖9為本發明所述Cdot-Au的熒光性能受激發光的影響,圖中標注為300nm,320nm,345nm,360nm,380nm,400nm的曲線分別表示對應激發波長下的熒光發射曲線(對應實施例2的產物);
圖10為本發明所述Cdot-Au的熒光穩定性,圖a為熒光強度受紫外光輻照的影響,圖b為熒光壽命,其中Cdot表示原料碳點,Cdot-Au表示Cdot-Au(對應實施例1和2的產物);
圖11為本發明所述Cdot-Au在不同原料比的制備條件下的紫外可見光吸收譜圖,圖中0.2∶1、0.4∶1、2∶1、3∶1、4∶1分別表示原料碳點與氯金酸的質量比(對應實施例3的產物);
圖12為本發明所述Cdot-Au的細胞毒性評估圖(MTT法)(對應實施例4);
圖13為本發明所述Cdot-Au的熒光細胞成像圖,a表示Cdot-Au與MCF-7細胞孵育6h后在熒光顯微鏡下的明場照片,b為Cdot-Au與MCF-7細胞孵育6h后熒光成像照片,c為Cdot-Au與CAL-27細胞孵育6h后在熒光顯微鏡下的明場照片,d為Cdot-Au與CAL-27細 胞孵育6h后的熒光成像照片(對應實施例4)。
具體實施方式
下面結合具體實施例對本發明進行詳細說明。以下實施例將有助于本領域的技術人員進一步理解本發明,但不以任何形式限制本發明。應當指出的是,對本領域的普通技術人員來說,在不脫離本發明構思的前提下,還可以做出若干變形和改進。這些都屬于本發明的保護范圍。
實施例1:碳點的制備
稱取2.0g一水合檸檬酸和1.0g L-半胱氨酸,加入10.0mL去離子水,攪拌至均勻混合;將上述混合溶液轉移至水熱反應釜中,于200℃馬弗爐中反應3h,自然冷卻至室溫,獲得棕黃色的碳點溶液。將所得碳點溶液經5000r/min離心,取上清液,用透析袋(截留分子量為3000Da)透析48h,蒸發干燥得純化的固態碳點,冷藏備用。
由圖1a碳點的TEM圖及圖2a粒徑分布圖可見,多數碳點粒徑為1~2.5nm,小于文獻報道的7nm粒徑,粒徑分布相當均勻,且呈單分散狀態。圖3a為碳點的紫外可見吸收光譜和熒光發射光譜圖,碳點在340nm波長處有最大吸收,在425nm波長處具有最大發射,表明碳點具有較大的斯托克位移,具有良好的紫外可見吸收和熒光性能。圖3b是激發波長從300nm逐漸增加到400nm時的熒光發射譜圖,碳點的最大發射波長位置并不受激發波長的移動而移動,而是固定在425nm處不變。取一水合檸檬酸的質量0.5g和3.5g時,所得碳點的性質與上述性質基本一致,將水熱反應溫度調整為180℃,反應時間調整為2h或8h,所得碳點的性能也基本保持不變。
本實施例中,一水合檸檬酸的用量可取0.5~3.5g中的任意值;L-半胱氨酸的用量可取0.5~2g中的任意值;反應溫度為180~200℃,反應時間為2~8h,離心速率為2000~5000r/min,透析袋的截留分子量為1000~8000Da,透析時間為12~48h。
實施例2:Cdot-Au的制備
1、原料碳點的制備
此實施例中碳點的制備方法如實施例1所示。
2、Cdot-Au的制備與表征
配制濃度為100μg/mL的氯金酸溶液,取5mL,緩慢滴加至5mL的一定濃度的碳點溶液中,保證碳點與氯金酸的質量比為3∶1,于60℃條件下,緩慢攪拌反應3h,用截留分子量為1000~5000Da的透析袋透析12~48h,即得到Cdot-Au納米復合粒子的水溶液,經過蒸發、干燥得到固體的Cdot-Au納米復合粒子。
Cdot-Au的形貌,以及與碳點的結構和性質的比較如下:
本發明所述的Cdot-Au在透射電鏡下為1~3nm左右的顆粒(圖1e及圖2e),粒徑大小與碳點的相似,且粒徑分布比較均一。根據X射線衍射(圖4)結果可知,碳點出現以2θ=19.98°為中心的寬衍射峰,而Cdot-Au的譜圖中,碳點的衍射峰位置偏移至2θ=22.21°,金納米團簇出現一個2θ=31.25°寬衍射峰,未見典型明顯的石墨和金顆粒衍射峰,這可能是由于碳點和金納米粒子為無定形結構或粒徑太小。由此可見,本方法制備的碳點為無定形的,與文獻報道不同。X射線光電子能譜(圖5)測試表明復合納米粒子上有金顆粒生成,并且碳點表面的官能團組成有明顯變化。在空氣氣氛下熱重分析(圖6)結果可知,Cdot-Au和碳點在200℃之前均有近10%的失重,對應于吸附水脫除過程;在250℃處具有明顯的熱解峰,但是Cdot-Au比碳點裂解更快,原因可能是高活性的金顆粒促進了樣品的裂解;560℃處的裂解峰則對應于碳點中類石墨結構的裂解;Cdot-Au在800℃條件下殘重約為9.9%,而此時碳點已經完全分解。Cdot-Au中金的質量百分比為9.9%,表明反應體系中幾乎所有添加的金前驅體均負載到碳點上。
為進一步研究Cdot-Au的化學組成,經比較Cdot-Au和碳點的紅外譜圖(圖7),發現Cdot-Au在3315~3545cm-1之間羥基伸縮振動吸收峰明顯減弱,表明了碳點表面的羥基在制備Cdot-Au中起到了還原作用。此外,1394cm-1處羧基離子的對稱伸縮振動峰也有明顯減弱,可能是羧基與金的相互作用影響了羧基離子的振動方式所致。巰基在2555cm-1的伸縮振動特征峰有明顯減弱但未完全消失,可能是制備碳點時有部分巰基被包埋到碳點中,無法與金原子結合。
通過紫外-可見光光譜和熒光光譜研究了Cdot-Au的紫外可見光吸收和熒光性質。如圖8所示,比較了碳點和Cdot-Au的紫外光譜。從圖8a可見,在365nm紫外光激發下,相同濃度的碳點和Cdot-Au均呈現明亮的藍色熒光;二者均在340nm處有最大紫外吸收峰,差別在于后者的紫外吸收有所降低。圖8b是Cdot-Au的紫外吸收和熒光發射圖,在最大吸收波長340nm處,其最大發射波長在425nm處,與碳點本身的紫外吸收和熒光性能相似。通過調節激發波長,考察了Cdot-Au的熒光性能是否受激發波長的依賴。如圖9所示,當激發波長在300~400nm范圍內變化時,Cdot-Au的熒光最大發射保持在425nm處,不隨激發波長的變化而移動,與碳點的激發獨立性相一致。以硫酸奎寧為參比,對Cdot-Au的量子產率進行了考察,量子產率計算公式為:
其中,表示待測樣品的量子產率,R表示參比樣品的量子產率,I表示待測樣品的熒光發射譜圖的積分面積,IR表示參比樣品的熒光發射譜圖的積分面積,A表示待測樣品在該濃度下的紫外吸收值,AR表示參比樣品在該濃度下的紫外吸收值,η是待測樣品溶劑的折射率,ηR是參比樣品溶劑的折射率。如表1所示,Cdot-Au的量子產率為34.7%,盡管相對于碳點的量子產率59%有所下降,仍是目前報道的量子產率最高的碳與金的復合納米材料。碳點的熒光來源于碳核骨架及其與表面官能團的相互作用。因而Cdot-Au的熒光性能下降的原因可能是在其制備過程中碳點表面的官能團發生了改變,如對熒光有極大貢獻的巰基與金生成Au-S共價鍵,而部分羥基則因還原金(III)離子而減少。
表1碳點和Cdot-Au的量子產率
通過檢測紫外光輻照不同時間后熒光強度的變化考察了Cdot-Au的熒光穩定性。由圖10a可見,經過波長為365nm紫外光分別輻照1、2、3、4、5h后,所有Cdot-Au的熒光發射曲線幾乎完全重合,熒光強度沒有出現明顯下降,表現出良好的耐光漂白性能;由圖10b可見,Cdot-Au熒光壽命為11.8ns,與碳點的熒光壽命11.5ns接近。實驗結果表明,Cdot-Au較好地保持了碳點的熒光性能,也間接證明金納米團簇在碳點表面的聚集并沒有顯著改變碳點的結構與光學性能。
實施例3:對Cdot-Au性能的調控
1、原料碳點的制備
此實施例中碳點的制備方法如實施例1所示。
2、碳點的用量對Cdot-Au的性能調控
通過調節碳點和氯金酸用量比考察了碳點用量對Cdot-Au的粒徑和光學性質的影響。如圖1b至圖1f及2b至圖2f所示,保持氯金酸濃度為50μg/mL條件下,當碳點與氯金酸的質量比為0.2∶1和0.4∶1時,復合納米粒子的粒徑約為35nm;當提高到2∶1時,得到兩種粒徑分布的納米顆粒,其中粒徑較大的約為9nm,較小的約為2nm;當提高到3∶1時,得到粒徑均勻的2nm左右的納米粒子,進一步提高到4∶1時,粒徑小于2nm。表明了Cdot-Au的粒徑隨碳點用量增加而逐漸變小,且分布均勻。由于碳點表面同時存在大量官能團,金陽離子(III) 首先與巰基和氨基通過化學鍵和靜電作用富集到碳點表面,再經過羥基或羧基將其原位還原成金原子,隨后金原子不斷聚集成為金納米團簇,由此可見,Cdot-Au的形成實際上是金元素異相成核的過程。因此,碳點在制備Cdot-Au時的成核、還原和穩定作用是否匹配是決定其粒徑的關鍵。當質量比小于2∶1時,還原和穩定作用不匹配,碳點表面生成金納米團簇后會繼續長大至粒徑為35nm以上;當碳點用量大于3∶1時,其還原和穩定作用匹配,Cdot-Au粒徑即使在室溫下30天仍能保持不變。
運用紫外可見吸收光譜考察了碳點和氯金酸質量比對紫外可見光吸收性能的影響。如圖11所示,當碳點與氯金酸的質量比在0.2∶1和4∶1區間變化時,Cdot-Au在540nm吸收峰由強變弱,而在340nm處碳點的吸收峰由弱變強。這是由于當質量比小時,形成了粒徑較大的金納米顆粒,導致其在500~600nm波長范圍內出現強吸收峰;質量比高時,主要生成附著在碳點的表面小于2nm的金納米團簇,而金納米團簇不會產生明顯的紫外吸收,因而復合納米粒子僅顯示出碳點的紫外特征吸收峰。
實施例4:Cdot-Au的毒性與生物成像
1、Cdot-Au的制備
此實施例中Cdot-Au的制備方法如實施例2所示。
2、Cdot-Au的細胞毒性評價
為考察本發明Cdot-Au的細胞毒性,利用MTT(噻唑藍)比色法,考察L929細胞(小鼠成纖維細胞)與碳點共同孵育48h后細胞的存活率(圖12)。用96孔板培養L929細胞,取10,50,100,200,400,500μg/mL的碳點溶液50μL,加入培養有L929細胞的孔中,孵化48h后,測定細胞的存活率。多次重復試驗表明,當碳點的濃度高達500μg/mL時,細胞的存活率仍然在70%以上,表明碳點對細胞的生長沒有顯著影響,具有很好的生物相容性。極小的細胞毒性是該復合納米粒子在生物上得以應用的基礎。
3、Cdot-Au在細胞成像上的應用
將Cdot-Au應用于人乳腺癌細胞MCF-7和人類口腔舌癌CAL-27的成像。利用6孔板培養上述細胞,加入50μL濃度為2mg/mL的Cdot-Au的溶液后共同孵育6h后,取出培養液,固定細胞后,封片,在熒光顯微鏡下觀察細胞。由圖13可見,與培養6h后,在紫外光(330~380nm)激發條件下,細胞的細胞質中可以發出明亮的藍色熒光,成像效果良好,而細胞核部分的熒光較弱,表明復合納米粒子僅進入細胞質,而沒有進入細胞核中。由于Cdot-Au沒有進行任何靶向修飾,不具有特異選擇性,根據文獻報道,Cdot-Au同樣可應用于其他細胞系的成像。如果在上引入合適的靶向修飾,則能實現亞細胞結構、細胞器或生物大分子的成像。
以上對本發明的具體實施例進行了描述。需要理解的是,本發明并不局限于上述特定實施方式,本領域技術人員可以在權利要求的范圍內做出各種變形或修改,這并不影響本發明的實質內容。
- 還沒有人留言評論。精彩留言會獲得點贊!