防粘連材料的制作方法
本發明涉及一種防粘連材料。特別涉及由水溶性高分子及脂肪族酯構成的固態或半固態基體、與作為該基體所含抗氧化物質的由抗壞血酸或其衍生物構成的防粘連材料。
背景技術:
:本專利申請人以提供防粘連材料為目的,在專利文獻1(日本專利第5686297號)公開了新穎的防粘連材料的發明,其中,于活體內,(a)貼附于活體組織時的分解性及(b)防粘連性能均優異,且較已知(c)濕潤時的處置性及(d)貼附于活體組織時的密接性均更優異。專利文獻1中記載的防粘連材料的主旨為,于基體層中使用水溶性高分子,且在該基體層的至少一面或雙面上配置由含活體分解性聚脂肪族酯的極薄膜(使用分光式橢圓偏光儀依波長380nm~900nm測定時,上述被覆層的光學厚度為27nm以上且小于160nm)構成的被覆層。另一方面,專利文獻2中公開有:以海藻糖為有效成分,且含有作為抗氧化物質的抗壞血酸衍生物的防組織粘連液的發明。[
背景技術:
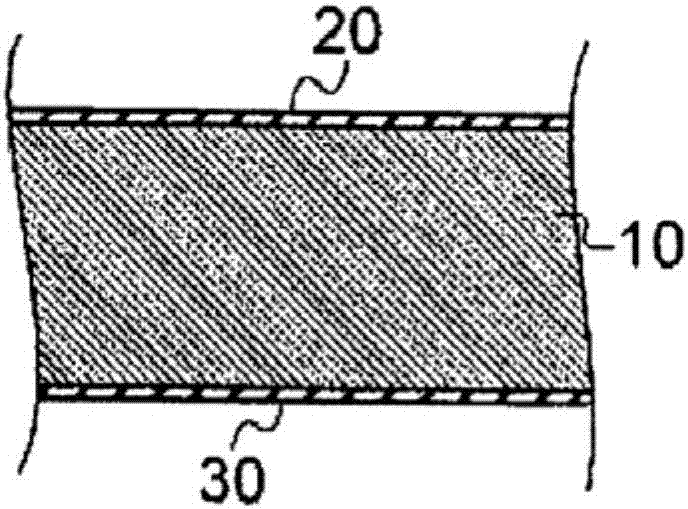
文獻][專利文獻]專利文獻1:日本專利第5686297號(權利要求書、圖1至圖4)專利文獻2:日本專利第4447640號(權利要求書、權利要求1至3)技術實現要素:(發明所要解決的問題)專利文獻1中記載的發明,雖為在活體內(a)貼附活體組織時的分解性、(c)濕潤時的處置性及(d)貼附于活體組織時的密接性等數方面而言提供具優異特性的防粘連材料的發明,但經本發明者等更進一步研究后,發現就確保更充分(b)防粘連性能方面、或以提供更優異防粘連材料為目的方面而言,尚殘留有數個待獲改善的課題。專利文獻2中記載的組織防粘連液屬于噴霧方式,就劑形而言,不同于專利文獻1中記載的薄膜或薄片狀防粘連材料,結果防粘連功能的表現機制便有所不同。(解決問題的技術手段)于是,本發明者為解決上述課題,經深入鉆研,結果達成下述發明。[1]根據本發明所提供的防粘連材料,在由水溶性高分子及聚脂肪族酯構成的固態或半固態基體中,含有抗壞血酸或抗壞血酸衍生物作為抗氧化物質。[2]再者,根據本發明所提供的防粘連材料,在[1]記載的防粘連材料中,上述固態基體為從薄膜狀、薄片狀及篩網狀的任一者中選擇的形態;上述薄膜狀、上述薄片狀及篩網狀的形態為,實質由該基體形成的單層構造、或具有在該基體的基體層上更進一步設置由聚脂肪族酯構成的被覆層的層積構造;或上述篩網狀的形態具有復合體所構成的纖維狀構造,且其基重為0.8g/m2~830g/m2,該復合體為,將固態的水溶性高分子及聚脂肪族酯構成的基體形成為棒狀而得到。[3]再者,根據本發明所提供的防粘連材料,在[1]記載的防粘連材料中,半固態基體為包含膠質狀的凝膠狀形態,具有單層構造或層積構造,37℃的粘度為100pa·s至1,000,000pa·s。[4]再者,根據本發明所提供的防粘連材料,在[2]記載的防粘連材料中,在該基體的基體層上具有被覆層的層積構造以3層形成,該3層為,在該基體層的雙面上分別具有被覆層的雙面被覆層及基體層。[5]再者,根據本發明所提供的防粘連材料,在[4]記載的防粘連材料中,當對上述被覆層的厚度使用分光式橢圓偏光儀以波長380nm~900nm測定時,上述被覆層的光學厚度為50nm以上且小于7000nm。[6]再者,根據本發明所提供的防粘連材料,在[1]記載的防粘連材料中,相對于構成基體或基體層的基材料重量(質量),含有0.5(w/w)%以上~10(w/w)%以下的抗壞血酸及其衍生物。[7]再者,根據本發明所提供的防粘連材料,將[1]至[6]中任一項記載的防粘連材料浸漬于牛血漿時,其24小時后的ph為7.5~6.0。[8]再者,根據本發明所提供的防粘連材料,在[2]至[7]中任一項記載的防粘連材料中,上述被覆層具備有:在上述基體層的一側面上所配置的第1被覆層、及在上述基體層的另一側面上所配置的第2被覆層,上述第1被覆層與第2被覆層為由同一材料料所構成。[9]再者,根據本發明所提供的防粘連材料,在[2]至[7]中任一項記載的防粘連材料中,上述被覆層具備有:在上述基體層的一側面上所配置的第1被覆層、及在上述基體層的另一側面上所配置的第2被覆層,上述第1被覆層與上述第2被覆層由不同材料所構成。[10]再者,根據本發明所提供的防粘連材料,在[1]至[9]中任一項記載的防粘連材料中,上述水溶性高分子為多糖類、蛋白質或合成高分子。[11]再者,根據本發明所提供的防粘連材料,在[1]至[10]中任一項記載的防粘連材料中,構成上述基體的水溶性高分子(a)與聚脂肪族酯(b)的重量比例ω(=a/b)為1~99/99~1。[12]再者,根據本發明所提供的防粘連材料,在實質由水溶性高分子構成的固態或半固態基體中,含有抗壞血酸或抗壞血酸衍生物作為抗氧化物質,或者更進一步在該基體的基體層雙面上分別具有由聚脂肪族酯構成的被覆層。[13]再者,根據本發明所提供的防粘連材料,在[2]至[12]中任一項記載的防粘連材料中,上述[1]的抗氧化物質含有屬于植物的脂溶性色素的類胡蘿卜素類或含多酚類的源自植物的抗氧化物質。(發明的效果)本發明的防粘連材料的特征在于,具有下述構成:基本上在由水溶性高分子及聚脂肪族酯構成的固態或半固態基體中,含有抗壞血酸或抗壞血酸衍生物作為抗氧化物質。本發明防粘連材料通過具有該構成,而達成下述事項:<1>由聚脂肪族酯及水溶性高分子構成的基體發揮作為創傷面與創傷面的物理性屏障(physicalbarrier)的功能,且利用連續地緩釋于創傷面的抗壞血酸或其衍生物促進創傷面治愈,由此,高效地表現出具有疊加效應的防粘連功能。<2>再者,基體形狀為具有固態(薄膜狀、薄片狀、篩網狀)或半固態(包含膠質狀的凝膠狀形態),且使此種形狀的基體或基體層中含有抗壞血酸或抗壞血酸衍生物,因而可確保上述物理性屏障功能,且能可靠地在該創傷面上表現出促進創傷面治愈效果。基體或基體層中所含有的抗壞血酸等會與形成具粘性溶液的水溶性高分子一起逐漸溶出,并滯留于基體表面(創傷面附近),而對促進治愈效果產生貢獻。<3>再者,在固相或半固態基體中調配抗壞血酸或抗壞血酸衍生物而使用的優點,為抗壞血酸可對該創傷面持續地緩釋。一般而言,創傷治愈通常至少24~48小時,依情況也會需要1~2周,在該所需期間內,抗壞血酸或抗壞血酸衍生物能連續地緩釋于創傷面而呈持續存在的狀態。另外,專利文獻1中記載的防粘連材料,過度在意加速基體層的分解吸收性,并未考慮這些抗壞血酸或抗壞血酸衍生物的緩釋,即便添加作為防粘連材料成分(創傷治愈促進成分)的抗壞血酸或抗壞血酸衍生物的情況,雖經足夠期間,但該抗壞血酸或抗壞血酸衍生物仍無法緩釋,會有無法獲得充分防粘連效果的可能性。即,專利文獻1中記載的防粘連材料中,基體層為由水溶性高分子構成,由于該基體層在水中較快速溶解并被分解吸收,因而沒有從該基體層緩釋抗壞血酸的考慮余地。再者,如專利文獻2中記載的組織防粘連液那樣,為呈液體狀,同樣地無法考慮藥劑緩釋、藥劑持續地緩釋于創傷面,不同于本發明所產生的防粘連功能的表現機制。關于此點,經本發明者等深入鉆研,結果確認到依后述代表例所表示的適當形狀的情況,能確保最佳的緩釋性。附圖說明圖1為用于說明本發明代表實施形態的防粘連材料1的圖,圖1(a)為防粘連材料1的立體圖,圖1(b)為其一部分的放大剖視圖。圖2為在防粘連材料1的基體層所添加的抗壞血酸的緩釋的隨時間變化評價圖。具體實施方式(定義)為了明確地說明本發明的防粘連材料,針對本發明的“基體”、“基體層”、“被覆層”設置以下定義。所謂“基體”為構成防粘連材料主要的基質部件,由水溶性高分子(a)及聚脂肪族酯(b)構成,于其中添加并含有抗壞血酸或抗壞血酸衍生物(以下有時稱為“抗壞血酸等”),由此構成防粘連材料。該防粘連材料置于活體內時,該基體在一定期間內保持形狀的狀態下緩釋抗壞血酸等。具體而言,該基體為了在活體內貼附于臟器,通常形成單層構造或層積構造的層構造,尤其將形成單層構造時的基體稱為“基體層”,又,在層積構造中具備有以被覆該“基體層”表面(雙面或單面)的方式所形成的外層(稱為“被覆層”)。通常,基體層為相對較厚的層,相較于基體層,被覆層構成為較薄的層。“被覆層”為如后述那樣的層,主要為控制來自基體層的抗壞血酸等的緩釋,又,當強度上保持基體層使防粘連材料置于活體中時,具有保持形狀的功能。根據上述,所謂“基體”屬于添加抗壞血酸等的基質部件,且為由此緩釋該抗氧化劑的物質,因而不僅“基體層”,就連“被覆層”也對應于“基體”(參照后記實施例5-6)。又,本發明的防粘連材料的基質部件,根據定義由水溶性高分子(a)及聚脂肪族酯(b)構成,但當為單層構造的情況,該層由水溶性高分子(a)及聚脂肪族酯(b)的雙成分組成物(a/b)所構成,另一方面,當為層積構造的情況,可設為例如典型地由“基體層”與“被覆層”所構成,則“基體層”實質上僅由水溶性高分子(a)構成,“被覆層”實質上僅由聚脂肪族酯(b)構成(參照后記實施例1-4)。以下,針對本發明的防粘連材料,根據圖1所示代表實施形態進行說明。但是,本發明的實施形態并不局限于圖1所示實施形態,在具備本發明特征的核心發明特定事項的前提下,可為各種實施形態。首先,針對本發明概要進行說明,本發明的防粘連材料在使由水溶性高分子及聚脂肪族酯構成的所謂固態基體中,含有抗壞血酸或抗壞血酸衍生物(以下有時稱為“抗壞血酸等”)。防粘連材料的形態,具體而言,固態基體為從薄膜狀、薄片狀及篩網狀中選擇的任一者,半固態基體為包含膠質狀的凝膠狀形態。當固態基體為薄膜狀、薄片狀的情況,其構造基本上為由單層構造所構成的基體層,但視所需也可如下述,設為在基體層上更進一步具備被覆層的層積構造。即,此種層積構造的情況,基體最典型地由:由水溶性高分子構成的基體層、及形成于其上面且由聚脂肪族酯構成的被覆層所構成。另外,一般而言,薄膜狀與薄片狀二者并沒有明確區分,本發明中將厚度小于200μm的情況設為薄膜狀,而薄片狀為設定為厚度達200μm以上的情況。篩網狀的情況為復合體所構成的纖維狀構造體,且其基重設定為0.8g/m2~830g/m2范圍內,該復合體為,將固態的由水溶性高分子與聚脂肪族酯構成的基體形成為棒狀而得到。當固態基體為半固態的情況,便為包含膠質狀的凝膠狀形態,具有單層構造或層積構造,且其在37℃下的粘度為100pa·s至1,000,000pa·s,該范圍的物性能表現出有效的物理性屏障功能。任一形態的基體均為由水溶性高分子及脂肪族酯構成的所謂固態或半固態基體,含有抗壞血酸等,這些的基礎在于,成為創傷面與創傷面的物理性屏障,以及有效率地可靠發揮緩釋于該創傷面的抗壞血酸等促進創傷面治愈所產生的疊加防粘連功能。[代表實施形態說明]圖1所示為用于說明本發明代表實施形態的防粘連材料1的圖。本發明并不局限于下述代表實施形態。雖未圖示,也可設為僅在如專利文獻1的圖2所示基體層單面上形成被覆層的情況,或者由在如圖4所示基體層的雙面上形成被覆層,再將其形成為棒狀復合體所構成的纖維狀(篩網狀)構造形態。圖1(a)為防粘連材料1的立體圖,圖1(b)為防粘連材料1的一部分的放大剖視圖。另外,圖1(a)及圖1(b)中,為求輕易理解發明,基體層10的層厚、以及第1被覆層20與第2被覆層30相對于基體層10的層厚,有某程度夸張的表示。代表實施形態的防粘連材料1為如圖1所示,具備有:由薄膜狀構成的基體層10、視需要設置在基體層10的一側面上所配置的第1被覆層20、以及在基體層10的另一側面上所配置的第2被覆層30。(基體的基本構成)本發明防粘連材料的特征在于:當基體為單層構造的情況,便由水溶性高分子(a)與活體分解性聚脂肪族酯(b)構成的二成分樹脂組合物(樹脂的均勻混合物)所構成;當層積構造的情況,便具備有:由水溶性高分子(a)構成的基體層、與由活體分解性聚脂肪族酯(b)構成的固態或半固態的被覆層。針對此點而言,徹底不同于基底為僅由水溶性高分子(單一成分)構成的基體層的專利文獻1所公開的已知產品。由于本發明的基體為由水溶性高分子與活體分解性聚脂肪族酯構成的二成分組合物的單層構造,或由上述層積構造構成,因而當其設置于活體中時,聚脂肪族酯部分(不同于水溶性高分子部分)不會迅速分解,能在相當長期間保持層狀(薄膜狀或薄片狀)形態(薄膜形狀保持部分)。另一方面,因為水溶性高分子部分會與屬于形狀保持部分的活體分解性聚脂肪族酯相互混合、或者相互接觸存在(不會如單一成分層的情況那樣比較迅速溶解),而從該基體表面接觸到活體液的部分逐漸溶出。如此從基體逐漸溶出(緩釋性)的水溶性高分子成分,可達對創傷面促進治愈的作用。(抗氧化物質/抗壞血酸等)本發明防粘連材料的特征在于:在如此所構成的二成分組合物構成的單層構造基體、或由上述層積構造構成的基體中,含有抗壞血酸或抗壞血酸衍生物作為抗氧化物質。根據本發明者等的研究,當在基體中調配抗壞血酸等,企圖從該基體緩釋時,如專利文獻1所記載,僅由聚三葡萄糖等水溶性高分子構成的基體層的情況,如上述,基體層自體在較短時間便會溶解,無法達成作為基質層(形態保持層)的功能,因而無法實現充分的緩釋性。另一方面,本發明中由水溶性高分子與聚脂肪族酯構成的二成分組合物所構成的單層構造基體、或層積構造基體的情況,活體分解性聚脂肪族酯可達基質(骨架形成)的功能,所以基體能長時間保持其形態。另一方面,本發明在基體中所調配的抗壞血酸等,會從基體表面溶出,由該二成分組合物構成的單層構造基體、或由層積構造構成的基體,如上述,例如水溶性高分子部分會與屬于形狀保持部分的活體分解性聚脂肪族酯相互混合,或者相互接觸存在,并從該基體表面逐漸溶出。該溶出的水溶性高分子部分,被認為會與(該部分所含有)抗壞血酸等一起溶出。如此,從基體溶出的水性高分子成分,便一起與抗壞血酸等逐漸溶出,并經常存在于創傷面,所以可認為達較高的治愈促進作用。(緩釋性的控制)本發明在基體中所含有抗壞血酸等的緩釋機制可認為如上述,因而抗壞血酸等的緩釋速度也可廣范圍控制。即,由水溶性高分子(a)與活體分解性聚脂肪族酯(b)構成的二成分組合物所構成的單層構造基體、或層積構造中,水溶性高分子(a)的比例越大,則該水溶性高分子的溶出量(溶出速度)越大(但是,若水溶性高分子的比例過大,則由于活體分解性聚脂肪族酯(b)減少,因而薄膜狀的形態保持力會降低)。另一方面,若增加聚脂肪族酯(b)的比例,則薄膜狀等基體的形態可長時間穩定地保持,相反,水溶性高分子(a)、以及抗壞血酸等的溶出速度會降低。若考慮以上事項,可配合其目的實現最佳的緩釋性。由此項觀點,本發明中,構成上述基體的水溶性高分子(a)與聚脂肪族酯(b)的重量比例ω(=a/b)較理想為1~99/99~1、更優選ω(=a/b)為20~80/80~20、特別優選ω(=a/b)為30~70/70~30。若水溶性高分子(a)的比例過少,則其溶出速度變小,且所調配的抗壞血酸等的緩釋速度極小。另一方面,若聚脂肪族酯(b)的量過大,則雖形狀保持性獲提升,但水溶性高分子(a)與抗壞血酸等的緩釋性變為過低。本發明的防粘連材料為當單層構造的情況,最有代表性的為,基體由水溶性高分子與活體分解性聚脂肪族酯所構成,當層積構造的情況,基體層10為如上述,由水溶性高分子所構成,而被覆層為由聚脂肪族酯所構成。首先,針對水溶性高分子進行說明。(水溶性高分子)水溶性高分子較優選為可使用多糖類、蛋白質或合成高分子。多糖類可適當使用例如:淀粉、直鏈淀粉、支鏈淀粉、糖原、聚葡甘露糖、糊精、葡聚糖、果聚糖等動植物儲存多糖類;纖維素、果膠、幾丁質等動植物構造多糖類;鹿角菜膠、瓊脂糖等源自海藻的多糖類;聚三葡萄糖等微生物多糖類;刺槐豆膠、瓜爾豆膠等植物膠多糖類;肝素、玻尿酸、硫酸軟骨膠、硫酸乙酰肝素、硫酸皮膚素、硫酸角質素等糖胺聚糖;以及這些多糖類的衍生物。蛋白質可適當使用例如:明膠、酪蛋白、膠原等。合成高分子可適當使用例如:聚乙烯醇、聚乙烯醇衍生物、聚丙烯酸類水溶性聚合物、聚丙烯酰胺、聚丙烯酰胺衍生物、聚環氧乙烷、聚環氧乙烷衍生物、聚乙烯吡咯啶酮、聚乙烯吡咯啶酮衍生物、聚酰胺類聚合物、聚環氧烷類聚合物、聚醚二醇類聚合物、順丁烯二酸酐共聚物類聚合物等。從提高防粘連材料全體柔順度的觀點而言,就所例示的水溶性高分子中,特別適合使用聚三葡萄糖。(聚脂肪族酯)接著,針對聚脂肪族酯進行說明。聚脂肪族酯可適當使用例如:聚(交酯)類;聚(乙交酯)類;聚(交酯-ε-乙交酯)類;聚(乳酸)類;聚(甘醇酸)類;聚(乳酸-ε-甘醇酸)類;聚己內酯類;聚酯酰胺類;聚酸酐類;聚原酸酯類;聚氰基丙烯酸酯類;聚醚酯類;聚(二氧雜環己酮)類;聚(亞烷基烷基化物)類;聚乙二醇與聚原酸酯的共聚物、以及其它這些的共聚物;高分子摻合物等。特別為從活體內適合性與活體分解性優異的觀點,較優選為使用聚(乳酸)類、聚(甘醇酸)類、聚己內酯類、及這些的共聚物中的至少一種。特別優選為乳酸/甘醇酸/ε-己內酯的三元共聚物(la/ga/ε-clt),分子量20,00~300,000左右者較適合。(基體層的厚度)基體或基體層10(以下有時簡稱為基體層10)的層厚,設定為例如1μm~5000μm。薄膜狀防粘連材料的情況,基體層10的層厚為小于200μm、較優選為1μm~150μm、更優選為10μm至100μm、特別優選為30μm~80μm。薄片狀防粘連材料的情況,基體層10的層厚形成為200μm以上。較優選為200μm至5000μm、更優選為300μm~3000μm、特別優選為500~2000μm、最優選為800~1000μm。基體層10的厚度為利用卡尺、微量測儀等直接接觸進行測定,此外,也可采用紅外線膜厚計、靜電容式厚度計、激光位移傳感器等適當儀器進行測定。(抗壞血酸、抗壞血酸衍生物)本發明中抗氧化成分的使用是為了更好地使抗壞血酸或其衍生物從防粘連材料緩釋,而使其含于基體或基體層(或視情況也可進一步含于被覆層)中。抗壞血酸(ascorbicacid)為以營養素維生素c的形式作用,具內酯構造有機化合物的一種,化學式為如下所示。在(iupac命名法)中視同呋喃的衍生物,表示為(r)-3,4-二羥基-5-((s)-1,2-二羥乙基)呋喃-2(5h)-酮。分子量為176.13g/mol、熔點為190℃、沸點為553℃的光學活性化合物。本發明可適當使用已知作為維生素c的l體、即l-抗壞血酸。[化1]本發明中,其它可使用的抗壞血酸衍生物,為包括有:抗壞血酸鈣、抗壞血酸鈉、磷酸-l-抗壞血酸鈉、磷酸-l-抗壞血酸鎂、抗壞血酸葡萄糖苷、抗壞血酸乙酯等。抗壞血酸或抗壞血酸衍生物,相對于防粘連材料全體重量(質量),較理想至少添加0.1(w/w)%以上~20(w/w)%以下、通常添加為0.5(w/w)%以上~10(w/w)%以下、較優選為1.0(w/w)%以上~9.0(w/w)%以下、更優選為1.5(w/w)%以上~8.0(w/w)%以下。將抗壞血酸或抗壞血酸衍生物依上述比例添加于基體或基體層,而使含有的本發明防粘連材料浸漬于100×100mm/30ml牛血漿時,經24小時后其ph成為7.5~6.0。其為用于促進創傷面治愈的最佳ph。此現象意味著當將本發明防粘連材料配置于活體內時,在該基體或基體層中所含有抗壞血酸等持續緩釋的前提下,防粘連材料配置部分附近的ph并不會偏離初期值,而是仍可維持治愈的最佳7.5~6.0值。如此,本發明防粘連材料的緩釋性為可配合目的而廣范圍變化,且可使創傷面附近長期間維持一定所需的ph。若針對此點更進一步演繹,則本發明中,若水溶性高分子部分從基體表面逐漸溶出,則抗壞血酸等也會一起溶出。值得一提的是,所溶出屬于水溶性高分子的聚三葡萄糖等會形成具粘性的溶液,因而推定該溶液會在溶解抗壞血酸等的狀態下,依覆蓋基體表面的方式滯留于該位置處。而且,具粘性的水溶性高分子溶液中的抗壞血酸等的濃度,理應大致等于基體中的濃度cm*(基體中調配cm*=8%時,合理認為溶出水溶性高分子溶液中的濃度也大致成為8%)。如此,含有抗壞血酸等的聚三葡萄糖等粘性溶液,會穩定地覆蓋創傷部,因而認為該創傷部為利用抗壞血酸與聚三葡萄糖等的疊加效應被保護,由此促進防粘連效果。本發明基體的形成為可將既定的抗壞血酸等樹脂組合物利用薄膜擠出法、片材料擠出法(sheetextrusion)形成,又也可從該樹脂組合物的溶液利用澆鑄法等形成。(其它抗氧化物質)本發明中,雖抗壞血酸等最具效果,但視所需也可取代抗壞血酸等,改為使用例如以下的抗氧化劑。即,可使用:維生素e;α-胡蘿卜素、β-胡蘿卜素、γ-胡蘿卜素、茄紅素、葉黃素等屬于植物脂溶性色素的類胡蘿卜素類;類黃酮素、兒茶素、單寧、花青苷、異黃酮、槲黃素等植物的花或葉、樹皮、莖等所含的源自含多酚類植物的抗氧化物質(sod用物質)等。(被覆層)本發明基本上為如上述那樣由水溶性高分子(a)與聚脂肪族酯(b)的組合物所構成的基體實質上形成的單層構造,即便未設置被覆層仍可達抗壞血酸等的緩釋性。其中,視所需,也可設為具有在該基體所構成的基體層上更進一步具有由聚脂肪族酯構成的被覆層的層積構造。此種層積構造的情況如前所述,最典型的情況為基體實質上由以水溶性高分子所構成的基體層、以及在其上所形成且由聚脂肪族酯構成的被覆層所構成。此情況下由聚脂肪酸酯所構成的被覆層的功能,基本上為控制添加于由水溶性高分子所構成的基體層的抗壞血酸的緩釋性。當被覆層不存在的情況,短時間內與水溶性高分子一起,抗壞血酸也會在短時間內溶出,而較難在充分的時間內確保其緩釋性。即,層積構造的情況,如圖1所示,可在基體層10的一面上形成被覆層20(第1被覆層),并在另一面上形成被覆層30(第2被覆層)。(被覆層的構成成分)作為被覆層的第1被覆層20及第2被覆層30,較優選為均由活體分解性聚脂肪族酯所構成。即,可使用與屬于基體構成成分的聚脂肪族酯相同的成分。為求謹慎,不厭其煩地再度進行例示。雜脂肪族酯可適當使用例如:聚(交酯)類、聚(乙交酯)類;聚(交酯-ε-乙交酯)類;聚(乳酸)類;聚(甘醇酸)類;聚(乳酸-共-甘醇酸)類;聚己內酯類;聚酯酰胺類;聚酸酐類;聚原酸酯類;聚氰基丙烯酸酯類;聚醚酯類;聚(二氧雜環己酮)類;聚(亞烷基烷基化物)類;聚乙二醇與聚原酸酯的共聚物、以及其它這些的共聚物;高分子摻合物等。特別為與構成基體層的情況同樣地,從活體內適合性與活體分解性優異的觀點,較優選為使用聚(乳酸)類、聚(甘醇酸)類、聚己內酯類、及這些的共聚物中的至少一種。更優選為上述特佳的乳酸/甘醇酸/ε-己內酯的三元共聚物(la/ga/ε-clt)。另外,代表實施形態中,第1被覆層20與第2被覆層30較優選為由這些例示材料中利用相同的材料所構成。(被覆層也含有抗壞血酸的情況)另外,基本上,被覆層由這些聚脂肪族酯所構成,但如后記當以該被覆層的強度保持為目的的情況,形成相對較厚的被覆層。于該情況,視所需也可使該被覆層含有抗壞血酸等。通過使其含于被覆層,特別為當將防粘連材料設置于創傷部時,在初期階段可可靠地執行緩釋。但由于被覆層的厚度ld如前述遠薄于基體層的厚度ld,因而該被覆層中所含抗壞血酸等的總量并無法如此增加,較難持續過長時間(長期間)緩釋,而最重要的為基體層中所含的抗壞血酸等便可達輔助性效果,故優選。單層構造僅于基體層添加抗壞血酸所形成,但該情況,由于抗壞血酸被包含于水溶性多醣類,因而其緩釋相對較快地進行。又,通過形成較薄的被覆層,如后記實施例3-4所記載的那樣,該抗壞血酸的溶出速度可實現適當地控制所需的緩釋率。另一方面,層積構造的情況如后記的實施例5-6,當形成較通常厚的被覆層的情況下,優選為于基體層與被覆層兩者添加抗壞血酸。當被覆層含有抗壞血酸的情況,可考慮依以下的方式進行被覆層部分的緩釋率(溶出速度)即,當將對被覆層的抗壞血酸的添加量(濃度cm*)設為一定,且改變被覆層的厚度(ld)的場合,在被覆層厚度ld越薄的初期階段中緩釋得越快,但在較短的時間內被覆層內的抗壞血酸匱乏。另一方面,被覆層厚度ld越厚緩釋越緩慢,但認為因被覆層內的抗壞血酸總量較多,而可持續長時間緩釋。又,若針對被覆層的厚度ld為一定,且改變對被覆層的抗壞血酸的添加量(濃度)cm*的情況進行考察,則在濃度cm*越高的初期階段中緩釋越快,濃度cm*越低則緩釋越緩慢。如上述那樣,通過適當調整被覆層的厚度ld、抗壞血酸的添加量(濃度)cm*,由于可任意地變更緩釋速度、緩釋持續時間等,因而可大幅地擴展治療方法的選項。對于被覆層添加的抗壞血酸的含有量(濃度)cm*,添加量為0.01(w/w)%以上~1.0(w/w)%以下、優選為0.02(w/w)%以上~0.8(w/w)%以下、更優選為0.1(w/w)%以上~0.5(w/w)%以下。(被覆層的厚度)被覆層的功能基本上如已記述那樣控制緩釋性(溶解速度)。例如后述實施例所示,通過設置由屬于生物分解性的聚脂肪族酯構成的被覆層,將其厚度控制于光學厚度為50nm以上且800nm以下,較優選為75nm以上且小于600nm左右的范圍內,便可使(來自基體的)抗壞血酸的緩釋性變化。換言之,本發明為如前述,基本上通過改變構成基體層的水溶性高分子(a)與聚脂肪族酯(b)的重量比例ω(=a/b),便可控制水溶性高分子(a)的溶出速度(及抗壞血酸等的溶出速度),更進一步通過形成被覆層,使其厚度(ld)在上述范圍內變化,便可更巧妙地進行緩釋性控制。被覆層(第1被覆層20與第2被覆層30)為使用分光式橢圓偏光儀,依波長380nm~900nm測定時的光學厚度,在基體層10中添加抗壞血酸或抗壞血酸衍生物的情況,為形成為50nm以上且800nm以下,較優選為75nm以上且小于600nm。較優選形成為75nm以上且500nm以下。即便被覆層(第1被覆層20與第2被覆層30)的光學厚度較薄(50~75nm左右),因為本發明在基體層10中有添加抗壞血酸等,故能發揮充分的防粘連性能。另外,若被覆層過厚(超過800nm),則活體中的被覆層分解性能會降低。又,由于來自基體層的抗壞血酸向活體中溶出(擴散)受到阻礙,因而從維持緩釋性的觀點而言并不優選。但,另一方面,從基體層(進而防粘連材料)的強度維持(形態保持)的觀點而言,要求被覆層本身具有不容易破裂程度的強度而具有某程度的厚度,例如后記實施例5-6所示,基本上設為800~7000nm、較優選為900~5000nm、更優選為1000~3000nm左右。如此,被覆層的厚度必需從強度維持的觀點考慮。即,如猴子或人類那樣的雙足步行動物,因腹腔內壓的上升或者胃或腸等臟器的移動,而有構成防粘連材料的基體(或被覆層)的薄膜狀或薄片狀的膜破裂的可能性。本發明通過后述的實施例5-6將猴子使用于動物實驗而重復研討,其結果可確認,上述程度的被覆層越厚越好。再者,如上述那樣,被覆層若過厚則有分解性能降低(抗壞血酸緩釋性降低)的危險,但當應用于如猴子或人類那樣的雙足步行動物,優先確保被覆層的機械強度的場合,基本上優選具有上述程度的厚度。此情況下,通過將調配于被覆層的抗壞血酸等的濃度cm*充分提高,可實質上抑制其緩釋性的降低。又,通過于相對較厚的被覆層中含有水溶性高分子,可某程度地抑制抗壞血酸緩釋性降低。基于上述,概括而言,被覆層的厚度ld形成為50nm以上且7000nm以下、優選為100nm以上且5000nm以下、更優選為1000nm以上且3000nm以下。而且,在此厚度范圍內,較優選為通過考慮防粘連材料所設置的腹腔內壓與力的環境、對象臟器的種類、所需緩釋期間等諸多條件,優先進行緩釋性、或優選進行形態保持性(但在任何情況下都必需考慮兩者),決定被覆層的厚度(及調配于被覆層中之抗壞血酸濃度cm*)。[實施例(a)]在如上述圖1所示的代表實施形態的防粘連材料1的構成基礎下,于基體層的材料中添加既定量l-抗壞血酸(和光純藥公司制),成形為薄膜狀的基體層雙面上,配置由相同材料構成的第1被覆層與第2被覆層(以下也有簡稱為“被覆層”的情況),如上所述,將ph調整為既定范圍的方式設為實施例試料(又,將未添加l-抗壞血酸的情況設為比較例試料)。(基體層的形成)具體而言,首先,基體層的材料為使用屬于水溶性高分子的聚三葡萄糖,在其中添加既定量抗壞血酸,利用澆鑄法對100mm×120mm×厚50μm的由聚三葡萄糖構成的膜進行成膜,而形成含抗壞血酸的基體層(基體層中的抗壞血酸濃度:cm)。(被覆層的形成)其次,依照常法的浸涂法施行被覆層的形成。即,制作經調整為既定濃度的屬于聚脂肪族酯的聚乳酸-聚甘醇酸-聚ε-己內酯的甲苯溶液(以下稱為涂布液),在該涂布液中浸漬上述調制的基體層,而在該基體層兩表面上形成由聚乳酸-聚甘醇酸-聚ε-己內酯三元共聚物構成的被覆層。經浸涂后在室溫中干燥30分鐘~1小時左右,將其設為實施例的試料(試驗片)。[比較例]除在上述實施例的基體層中未添加(未含有)抗壞血酸外,其余均與上述實施例同樣地制作防粘連材料,設為比較例的試料(試驗片)。(被覆層的光學厚度的測定方法)由于本發明的被覆層屬于極薄膜,因而優選以光學厚度(ld)表示。在基體層上所層積的被覆層的光學厚度(ld)的測定,為使用分光式橢圓偏光儀(j.a.woollamjapan公司制“alpha-se(美國注冊商標)”)。另外,測定波長設為380nm~900nm。(實施例1~3)(抗壞血酸添加濃度cm*的研究)將上述由聚乳酸-聚甘醇酸-聚ε-己內酯構成被覆層的厚度(ld)固定為300nm,改變對基體層的抗壞血酸的添加濃度cm(w/w)%,浸漬于牛血漿后,測定24小時后的ph(24)。結果示于表1。即,實施例1的抗壞血酸添加濃度cm=2.0(w/w)%、ph(24)=7.0、實施例2為cm=4.0(w/w)%、ph(24)=7.0,實施例3為cm=8.0(w/w)%、ph(24)=6.0。(比較例1)(無添加抗壞血酸的情況)比較例1為除了基體層中未添加抗壞血酸(cm=0%)外,其余均施行與實施例1~3同樣的試驗(被覆層的厚度(ld)=300nm)。浸漬于牛血漿后24小時后的ph(24)=7.3。結果示于表1。(實施例4)(被覆層的厚度(ld)的研究)除將被覆層的厚度(ld)設為75nm外,其余均施行與實施例3同樣的試驗。結果如表2所示,cm=8.0%的情況,與ld=300nm時的ph(24)=6.0之間差異并未被確認。(比較例2~3)(被覆層的厚度(ld)的研究)除將被覆層的厚度(ld)設為75nm(比較例2)或600nm(比較例3)外,其余均施行與比較例1同樣的試驗。結果與比較例1(ld=300nm)一并示于表2。在基體層中未添加抗壞血酸(cm=0%)的比較例的情況,即便使被覆層的厚度(ld)以75nm→300nm→600nm變化,仍為ph(24)=7.3(一定)。(抗壞血酸添加量與ph)[表1]比較例1實施例1實施例2實施例3被覆層的光學厚度ld(nm)300nm300nm300nm300nm抗壞血酸濃度cm(w/w)(%)0%2.0%4.0%8.0%浸漬牛血漿時的ph(24hr)7.37.07.06.0(被覆層的光學厚度與抗壞血酸添加量)[表2]比較例2比較例1比較例3實施例4被覆層的光學厚度ld(nm)75nm300nm600nm75nm抗壞血酸濃度cm(w/w)(%)0%0%0%8.0%浸漬牛血漿時的ph(24hr)7.37.37.36.0[試驗例i](防粘連性能評價)本試驗例1為用于評價防粘連材料的防粘連性能的試驗(另外,本試驗例1為對應于本案申請人所提出的專利文獻1的試驗例2)。在評價防粘連性能時,由實施例與比較例的試料制作試驗片,將試驗片貼附于豬的腹腔內,觀察粘連程度并評級,以評價防粘連性能。(1)試驗片的準備裁剪上述實施例1~4與比較例1~3的試料,而制作100mm×120mm長方形狀的試驗片。(2)試驗方法(粘連模型的制作)首先,將豬在全身麻醉下在腹部正中央切開15cm而剖開腹部,使小腸露出于創口外。其次,針對所露出的小腸的一定面積(1×5cm左右),使用銼刀摩擦直到出現點狀出血。在出現點狀出血后,正確地暴露于空氣中10分鐘。然后,將小腸塞回腹腔內,于切開部分的正下方貼附上述試驗片的防粘連材料。腹壁使用吸收性縫合線(2-0)依雙層連續縫合而封閉,以制作粘連模型。另外,實施例、比較例均設為各組5例左右的例數。(3)試驗方法(粘連程度的觀察、粘連評級的計算)從粘連模型制作起14天后,使豬在全身麻醉下放血而犧牲死亡,肉眼觀察剖腹正中央創口正下方的粘連,求取粘連的發生機率。另外,本件為屬于下腹部的評價,故忽視上腹部的肝臟粘連。試驗結果示于以下的表3、表4。表3與表4的讀法,為例如表3的“實施例1”(被覆層的厚度(ld)300nm、抗壞血酸添加濃度cm=2.0%)的欄,對應于表4的“粘連發生率σ:20%(1/5)”(被覆層的厚度(ld)300nm、抗壞血酸添加濃度cm=2.0(w/w)%的欄);而表3的“比較例2”(被覆層的厚度(ld)75nm、抗壞血酸添加濃度cm=0%)的欄,對應于表4的“粘連發生率σ:75%(3/4)”(被覆層的厚度(ld)75nm、抗壞血酸添加濃度cm=0%的欄)。[表3][表4](考察1)(1)(抗壞血酸添加濃度cm的研究(i))由表4可確認到當被覆層的厚度(ld)設為300nm的情況,改變對基體層的抗壞血酸既定添加濃度cm(2.0(w/w)%至8.0(w/w)%)的實施例1(粘連發生率σ為20%)及實施例2~3(粘連發生率σ為0%)的結果,對比于被覆層的厚度(ld)同為300nm而未添加抗壞血酸的比較例1(粘連發生率σ為66%),實施例的情況明顯降低粘連發生率。又,若將實施例1與實施例2~3進行比較,可確認到抗壞血酸的添加濃度cm從2.0(w/w)%增加至4.0~8.0(w/w)%,粘連發生率σ呈現20%→0%的更加降低狀態。(2)(抗壞血酸添加濃度cm的研究(ii))將被覆層的光學厚度(ld)固定為75nm,并對基體層高濃度(cm=8.0(w/w)%)添加抗壞血酸的實施例4(粘連發生率σ=50%),相較于未添加抗壞血酸的比較例2(粘連發生率σ=75%),可確認到粘連發生率σ降低。[試驗例ii]本試驗例ii為用于評價表現防粘連性能的抗壞血酸緩釋性的試驗。即,將對基體層的抗壞血酸添加濃度cm=8.0w/w%(一定),與僅被覆層的厚度ld不同的情況([實施例3](ld=75nm)及[實施例4](ld=300nm))進行比較,確認是否達成最佳的緩釋。試驗片的準備裁剪上述實施例及比較例的試料,制作50mm×50mm正方形狀的試驗片。試驗方法在磷酸緩沖液(ph7.4)100ml中,浸漬1片前述實施例3(ld=75nm)及實施例4(ld=300nm)的防粘連材料,測定磷酸緩沖液中的抗壞血酸濃度隨時間變化直到24小時為止。抗壞血酸的定量為采用使用維生素c定量套件(cosmobio公司制)的比色定量法實施。再者,將防粘連材料中所含抗壞血酸量全量溶出于磷酸緩沖液時的濃度設為100%,求取各測定點的緩釋率再者,試驗所使用的材料為24小時觀察形狀。(試驗結果)試驗結果示于圖2。另外,超過24小時的數據,因為抗壞血酸已分解故不采用。直到24小時為止的數據中,實施例3的緩釋率為60~70%,實施例4的緩釋率為20~30%左右。若比較二者,被覆層的厚度ld較薄的實施例4(ld=75nm),相較于被覆層的厚度較大的實施例4(ld=300nm),緩釋速度更大,在經過時間t=10hr時溶出70%以上。相對于此,得知具有更厚被覆層的實施例4(ld=300nm),緩釋更緩慢(即便t=24hr仍為左右)。然而,關于t=14天后的粘連發生率σ,實施例3的粘連發生率σ為0%,達遠較實施例4的50%更優越的防粘連效果。此現象推定為實施例3中經過t=14天仍維持最佳緩釋速度的緣故。反之,可認為實施例4的情況,為因為緩釋速度過大,導致在短時間內該基體層中的抗壞血酸耗盡的緣故。再者,試驗所使用的材料(試驗片)雖經24小時后幾乎凝膠化而維持形狀,但可謂呈半固態。(考察2)依照使用固態或半固態的材料,得知緩釋率會有差異。更具體而言,設置由屬于生物分解性的聚脂肪族酯構成的被覆層,且將其厚度控制于光學厚度75nm以上且小于600nm的范圍,便可長時間維持(添加于基體的)抗壞血酸的緩釋性。此現象表示可依照適用的損傷部位或損傷深度,調整材料特性。又,不會有如液性物(例如專利文獻2)般在剛貼附后便(全量短暫)緩釋,若根據本發明具有基體層形態的防粘連材料,可隨時間變化(長期間,例如實施例所示14天)仍維持緩釋、即長期間進行緩釋。[實施例(b)]與實施例(a)同樣地在如圖1所示實施形態的防粘連材料1構成基礎上,于基體層的材料中添加既定量l-抗壞血酸(和光純藥公司制),在成形為薄膜狀的基體層雙面上,同樣地配置添加l-抗壞血酸的第1被覆層及第2被覆層(以下也有同樣地簡稱為「被覆層」的情況),并將其設為實施例試料。(又,將于基體層、被覆層均未添加l-抗壞血酸的情況設為比較例試料。)(實施例5-6)(添加抗壞血酸后的較厚的被覆層的形成)(基體層的形成)與實施例(a)同樣,基體層的材料使用作為水溶性高分子的三葡萄糖,在其中(以成為cm=4(w/w)%的方式)添加既定量的抗壞血酸,利用澆鑄法對100mm×120mm×厚(ld)50μm的由聚三葡萄糖構成的膜進行成膜,以形成含抗壞血酸的基體層。(被覆層的形成)其次,依照浸涂法進行被覆層的形成。即,制作調整為既定濃度的作為聚脂肪族酯的聚乳酸-聚甘醇酸-聚ε-己內酯的甲苯溶液(以下稱為涂布液),在該涂布液中以成為既定量(cm*=0.4(w/w)%)的方式添加抗壞血酸。于該含抗壞血酸涂布液中浸漬上述調制的基體層,而在該基體層兩表面上形成由聚乳酸-聚甘醇酸-聚ε-己內酯三元共聚合體構成的被覆層。經浸涂后在室溫中干燥30分鐘~1小時左右,通過重復施行此涂布步驟(浸涂)、干燥步驟,而形成較實施例1-4更厚膜厚(ld)的被覆層(基體層雙面的第1外層、第2外層)(其中,cm*=0.4(w/w)%)。于此,形成被覆層的厚度ld=1000nm(實施例5)、ld=3000nm(實施例6)的試料(試驗片)。(比較例4)(無添加抗壞血酸的情況)再者,比較例與實施例5-6同樣,于基體層(ld=50μm)兩側形成被覆層(ld=1000nm),但于任一層中均完全未添加抗壞血酸(cm=0、cm*=0),將其設為比較例4的試料(試驗片)。[試驗例iii](防粘連性能評價)本試驗例iii用于評價與試驗例i同樣防粘連材料的防粘連性能的試驗,但將對象動物種類變更為更接近人類。該情況假設防粘連材料的應用環境(腹腔內壓或臟器移動的發生)為更嚴酷的條件,以確認是否適合此環境。在評價防粘連性能時,從上述實施例(b)及比較例的試料制作試驗片,將試驗片貼附于猴子(macacafascicularis)的腹腔內并觀察粘連的程度,且通過測定粘連長度,評價防粘連性能。(1)試驗片的準備裁剪上述實施例5-6及比較例4的試料,制作100mm×120mm長方形狀的試驗片。(2)試驗方法(粘連模型的制作)首先,將猴子在全身麻醉下在腹部正中央切開8cm而剖開腹部,使大腸露出于創口外。其次,針對所露出之大腸的一定面積(3×4cm左右),使用銼刀摩擦直到出現點狀出血。在出現點狀出血后,正確地暴露于空氣中10分鐘。然后,將大腸塞回腹腔內,于切開部分的正下方貼附上述試驗片的防粘連材料。腹膜、腹壁及肌肉層使用尼龍縫合線(3-0)連續縫合,皮下組織及皮膚利用尼龍縫合線(4-0)簡單結扎并封閉,以便制作封閉的粘連模型。另外,實施例5-6、比較例4均設為各組3例左右的例數。試驗方法(粘連程度的觀察、粘連長度比例的測定)從粘連模型制作起14天后,在猴子全身麻醉下開腹,肉眼觀察正中央創口正下方的粘連,求出粘連的發生機率、粘連長度比例。(考察3)從為了提高防粘連材料的強度且確實地施行形態維持的觀點來看,相對由與實施例1-4相同厚度的聚三葡萄糖構成的基體層(ld=50μm)(于其雙面)形成更厚的聚脂肪酸酯的被覆層的情況,通過形成該較厚的被覆層,認為可阻礙來自基體層的抗壞血酸的緩釋。然而,即便在該情況下(ld=1000nm(實施例5)、ld=3000nm(實施例6)),因該被覆層也含有抗壞血酸,故如表5所示,表示可使粘連發生率顯著降低。(再者,確認到所貼附的防粘連材料的形狀于14天后也幾乎保持當初的形態,且確認到通過具備實施例中所使用程度的厚度的被覆層,從形態維持的觀點而言,防粘連材料可達到初期之目的。)即,于實施例5中,當將被覆層的厚度(ld)設為1000nm的情況,相對于基體層的抗壞血酸濃度cm(4.0(w/w)%),通過將被覆層的抗壞血酸濃度cm設為0.4(w/w)%,可使粘連發生率σ降低為67%(2/3)、粘連長度比例β(=l*/l)降低為15.50%。這一情況的原因認為是,當將比較例4的相同被覆層的厚度(ld)設為1000nm的情況,相較于在基體層及被覆層中未添加抗壞血酸時的(濃度cm=0%、cm*=0%)粘連發生率σ為100%(3/3)、粘連長度比例β(=l*/l)為82.60%,認為可顯著提升抗粘連性能的評價。又,于實施例6中,雖將被覆層的厚度(ld)設為更厚的3000nm,但于此情況下也與實施例5同樣地通過將基體層的抗壞血酸濃度cm設為4.0(w/w)%、被覆層的抗壞血酸濃度cm*設為0.4(w/w)%,相較于比較例4,表示可使粘連發生率σ顯著地降低為33%(1/3)、粘連長度比例β(=l*/l)顯著地降低為33.30%。再者,進一步進行研討,若與表3的實施例2(基體(ld=50μm)的抗壞血酸濃度cm=4.0(w/w)%、被覆層(厚度ld=300nm,但抗壞血酸濃度cm*=0%)進行比較,粘連發生率σ在表4(cm=4.0%的情況)中為0%(無粘連),相對于此,于實施例5-6中粘連發生率σ并非完全沒有,可確認其差異。基本上,此差異的最大原因可認為,因供于試驗的動物種類(豬與猴子)不同,而使基線粘連的發生頻率不同,且為了將被覆層的厚度(ld=1000或3000nm)保持作為防粘連材料的強度,因而基于一直較厚地形成等理由而產生,并非源自防粘連材料的性能的本質差異。在此項觀點中,認為通過使添加于該被覆層的抗壞血酸濃度cm*進一步增加為0.4%→0.6%→0.8%→1.0%等,可充分減低粘連發生率σ。又,認為雖目前被覆層ld為僅由活體分解性的聚脂肪酸酯(b)構成,但通過于被覆層ld中含有少量的例如與抗壞血酸相同程度的聚三葡萄糖等的水溶性高分子(a),可采用更加速抗壞血酸緩釋等的手段,進一步減低粘連發生率。[表5]另外,關于專利文獻1中記載的下述試驗例1、試驗例3、試驗例4,也是為求謹慎而施行對應的試驗,但如下所述,得知實用上并沒有功能性問題,因而此處不記載詳細內容。即,(i)(試驗例1)(防粘連材料貼附于活體組織時的分解性評價):貼附于活體直到表現強度為止的時間雖稍長,但不致影響功能。(ii)(試驗例3)(防粘連材料濕潤時的處置性評價):試驗例2中,雖施行豬以及猴子腹腔內的貼附,但能毫無問題地貼附。(iii)(試驗例4)(防粘連材料貼附于活體組織時的密接性評價):試驗例2中,雖施行豬以及猴子腹腔內的貼附,但能毫無問題地貼附。(產業上的可利用性)本發明的防粘連材料為基本上使由水溶性高分子(a)與活體分解性聚脂肪族酯(b)構成的固態或半固態基體中,含有作為抗氧化物質的抗壞血酸或抗壞血酸衍生物的構成,因而由水溶性高分子與聚脂肪族酯構成的基體,發揮作為創傷面與創傷面的物理性屏障的功能,且通過緩釋的抗壞血酸或其衍生物促進創傷面的治愈,能效率極佳地表現出疊加效應的防粘連功能,故在醫療領域中產業上的可利用性極大。符號說明1防粘連材料10基體層20第1被覆層30第2被覆層cm基體層的抗壞血酸等的濃度cm*被覆層(外層)的抗壞血酸等的濃度ld基體層的厚度ld被覆層的厚度(光學厚度)l切開創口長度l*切開創口的粘連長度抗壞血酸的緩釋率(溶出速度)。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1