電子照相感光體以及使用其的圖像形成裝置及圖像形成方法與流程
本發明涉及電子照相感光體以及使用其的圖像形成裝置及圖像形成方法。
背景技術:
近年來,就電子照相方式的圖像形成裝置而言,隨著工序速度的高速化(曝光-顯影間時間的縮短化),要求進一步的高耐久性及高畫質化,對于圖像形成裝置具備的電子照相感光體也要求改進。作為針對于這樣的要求的技術,在專利文獻1及專利文獻2中公開有在保護層中含有p型半導體微粒的電子照相感光體。
現有技術文獻
專利文獻
專利文獻1:日本特開2013-130603號公報
專利文獻2:日本特開2015-175937號公報
技術實現要素:
發明要解決的課題
但是,在上述專利文獻1及2中所記載的技術中,雖然耐久性在某種程度上得到改善,但在高速的工序速度下的圖像形成中抑制圖像記憶(メモリ)、翳影(カブリ)的發生這樣的圖像性能的方面,要求進一步的提高。
因此,本發明鑒于上述的問題而完成,目的在于提供耐久性優異、以高速的工序速度進行圖像形成的情況下也使圖像記憶、翳影的產生減少了的電子照相感光體。
用于解決課題的手段
本發明人為了解決上述的問題,進行了深入研究,結果發現通過具有以下的構成的電子照相感光體能夠解決上述課題,完成了本發明。
1.電子照相感光體,其為在導電性支承體上至少依次層疊感光層及保護層而成的電子照相感光體,其中,
上述保護層含有包含p型半導體微粒、n型有機半導體及聚合性化合物的固化性組合物的固化物。
2.上述1.所述的電子照相感光體,其中,上述n型有機半導體含有選自由以下述通式(1)、(2a)、(2b)、(3a)、(3b)、(4)和(5)表示的化合物組成的組的至少1種的化合物:
【化1】
上述通式(1)中,
xa為由下述的化學式(1-1)~(1-5)的任一者表示的4價的基團,可具有選自由取代或未取代的c1~c8烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基及羥基組成的組的至少1個取代基,
【化2】
r11及r12各自獨立地選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基及硝基組成的組;
【化3】
上述通式(2a)及(2b)中,
xb為取代或未取代的c6~c18亞芳基,
a1及a2各自獨立地為亞噁二唑基,
r21及r22各自獨立地為取代或未取代的c6~c18芳基,其取代基選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組;
【化4】
上述通式(3a)中,
r301~r304各自獨立地為選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基所組成的組的基團,可相互連接而形成環結構,此時,上述環結構可以為芳香環或非芳香環的任一種,可具有至少1個選自由硫(s)、氮(n)和氧(o)組成的組的雜原子;
【化5】
上述通式(3b)中,
r305~r312各自獨立地為選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團,可相互連接而形成環結構,此時,上述環結構可以為芳香環或非芳香環的任一種,可具有至少1個選自由硫(s)、氮(n)和氧(o)組成的組的雜原子;
【化6】
上述通式(4)中,
q1~q6各自獨立地為碳原子或氮原子,
r41及r42各自獨立地為選自由氫原子、c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團,可相互連接而形成環結構,
r43及r44各自獨立地為選自由氫原子、c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、取代或未取代的c6~c18芳基、氰基、硝基、取代或未取代的c2~c24雜芳基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團;
【化7】
上述通式(5)中,
z為碳原子或氮原子,
r51及r52的至少一者為腈基、或者取代或未取代的c1~c8烷氧基羰基,
r53~r60各自獨立地為選自由氫原子、c1~c8烷基、取代或未取代的c6~c18芳基及取代或未取代的c1~c8烷氧基羰基組成的組的基團。
3.上述1.或2.所述的電子照相感光體,其中,上述p型半導體微粒為選自由下述化學式(6)~(8)表示的化合物組成的組的至少1種的化合物:
【化8】
m1cu2o2…(6)cum2o2…(7)cum3o…(8)
上述化學式(6)~(8)中,m1為第2族元素,m2為第13族元素,m3為第5族元素。
4.上述1.~3.的任一項所述的電子照相感光體,其中,上述p型半導體微粒在表面具有反應性有機基團。
5.上述1.~4.的任一項所述的電子照相感光體,其中,上述聚合性化合物具有(甲基)丙烯酰基。
6.圖像形成裝置,其具備上述1.~5.的任一者所述的電子照相感光體。
7.圖像形成方法,其中,使用上述6.所述的圖像形成裝置來形成電子照相圖像。
發明的效果
根據本發明,提供耐久性優異、即使在以高速的工序速度進行圖像形成的情況下也減少圖像記憶、翳影的發生的電子照相感光體。
附圖說明
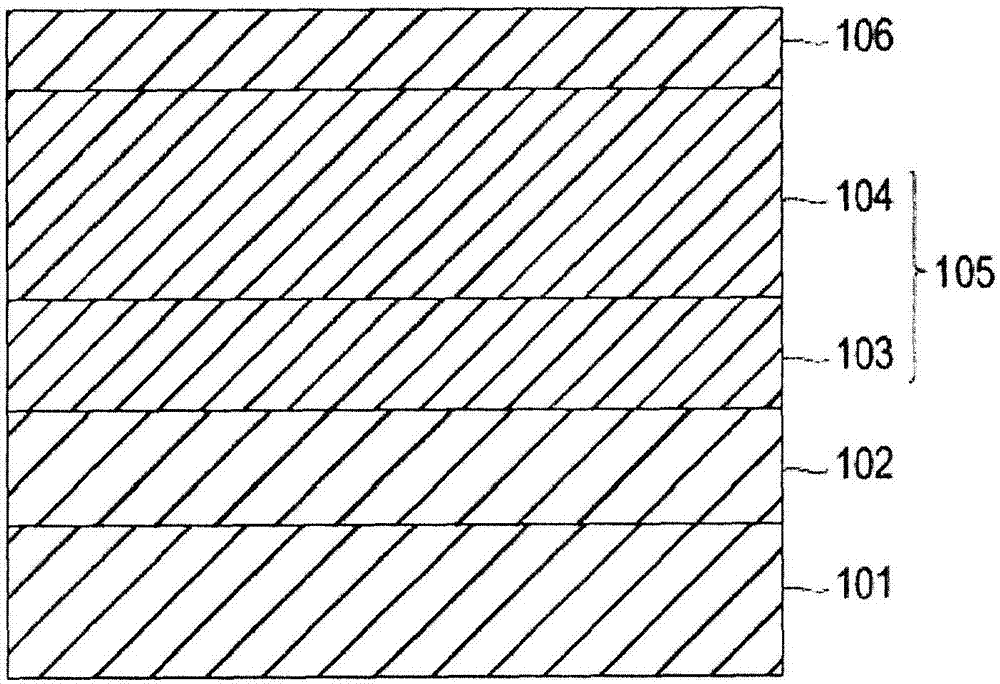
圖1為表示本發明的一實施方式的電子照相感光體的概略剖面圖。
圖2為表示本發明的一實施方式的彩色圖像形成裝置的概略剖面圖。
符號的說明
1,1y,1m,1c,1bk感光體
2y,2m,2c,2bk帶電手段
3y,3m,3c,3bk曝光手段
4y,4m,4c,4bk顯影手段
5y,5m,5c,5bk一次轉印輥
5b二次轉印輥
6y,6m,6c,6bk,6b清潔手段
10y,10m,10c,10bk圖像形成單元
20給紙盒
21給紙手段
22a,22b,22c,22d中間輥
23定位輥(レジストローラ)
24定影手段
25排紙輥
26排紙托盤
70中間轉印體單元
71,72,73,74輥
77中間轉印體
80殼體
82l,82r支承軌
101導電性支承體
102中間層
103電荷產生層
104電荷傳輸層
105感光層
106保護層
a主體
sc原稿圖像讀取裝置
p轉印材料
具體實施方式
以下,對本發明的實施方式詳細地說明。應予說明,本發明并不只限定于以下的實施方式。
應予說明,在本說明書中,表示范圍的“x~y”意味著“x以上且y以下”。另外,只要無特別記載,操作及物性等的測定是在室溫(20~25℃)/相對濕度40~50%rh的條件下進行測定。另外,本說明書中,“(甲基)丙烯酰基”這樣的術語是指甲基丙烯酰基及丙烯酰基。
另外,本說明書中,“取代”,只要無特別定義,是指被c1~c20烷基、c2~c20烯基、c2~c20炔基、c1~c20烷氧基、烷氧基羰基(-coor,r為c1~c20烷基)、鹵素原子(f、cl、br或i原子)、c6~c30芳基、c6~c30芳氧基、氨基、c1~c20烷基氨基、氰基、硝基、硫醇基、c1~c20烷硫基或羥基取代。
根據本發明的一方式,提供在導電性支承體上至少將感光層及保護層依次層疊而成的電子照相感光體。而且,該電子照相感光體的保護層在含有包含p型半導體微粒、n型有機半導體及聚合性化合物的固化性組合物的固化物的這點上具有特征。
根據本發明,提供耐久性優異、即使在以高速的工序速度進行圖像形成的情況下也使圖像記憶、翳影的產生減少的電子照相感光體。對于產生這樣的效果的機理尚不完全清楚,但推定以下的機理。
在專利文獻1及2中公開的電子照相感光體的情況下,作為用于應對圖像形成裝置的高速化的手段之一,有增加保護層中的p型半導體微粒的含量的手段。認為由此能夠減少圖像記憶的發生,但如果保護層中的p型半導體微粒的量增加,則微粒之間接觸的部位(概率)增加,形成局部的導通路徑。其結果,即使在帶電工序中將電荷給予感光體表面,導通部位附近的表面電位也局部地降低(沒有附帶電荷)。由此,產生調色劑向非曝光部的附著、發生所謂的翳影這樣的新的問題。
另一方面,就本發明涉及的電子照相感光體而言,在保護層中與p型半導體微粒一起還添加有n型有機半導體。通過該n型有機半導體的添加,對保護層賦予電子傳輸性和電荷傳輸性這兩者,在保護層內能夠使曝光后的殘留電位向電荷傳輸層的方向移動,來自電荷產生層的空穴(hole)容易向最外表面移動。因此,推測與以往的感光體相比,能夠容易地、迅速地消除殘留電位(負電荷)。目前為止也考慮過如何使來自電荷產生層的空穴迅速地移動至最外表面從而在最外表面使殘留電位消除,但在本發明中,基于使最外表面的殘留電位向感光層內部(電荷傳輸層側)移動、即、使電子從感光層的上層向下層移動這樣的新的體系設計,消除了表面電位的波動。由此,認為即使在以高速的工序速度進行圖像形成的情況下,也不會使p型半導體微粒增加,能夠減少圖像記憶、翳影的發生,得到具有優異的耐久性的電子照相感光體。另外,專利文獻2涉及的電子照相感光體使用金屬氧化物(氧化錫)作為n型半導體,而本發明涉及的電子照相感光體使用有機化合物作為n型半導體。認為:由此涂布液的分散性、制膜性提高,且良好地抑制翳影的發生。
予以說明,本發明并不受上述機理的任何限制。
以下,對本發明的電子照相感光體以及使用其的圖像形成裝置及圖像形成方法進行說明。
<電子照相感光體>
圖1為根據本發明的一實施方式的電子照相感光體的概略剖面圖。本實施方式涉及的電子照相感光體在導電性支承體101上依次層疊有中間層102、由電荷產生層103及電荷傳輸層104組成的感光層105、和保護層106。即,本發明涉及的電子照相感光體在導電性支承體上至少依次層疊感光層及保護層而成。
以下,對構成感光體的各層詳細地說明。
[保護層]
(保護層的構成材料)
本發明涉及的電子照相感光體的保護層含有包含p型半導體微粒、n型有機半導體及聚合性化合物的固化性組合物的固化物。以下對各成分進行說明。
《p型半導體微粒》
p型半導體微粒是指使用空穴(hole)作為傳輸電荷的載流子的半導體微粒。
就本發明的保護層中所含的p型半導體微粒而言,從空穴傳輸能力及實用性的觀點出發,優選為含有cu元素的氧化物,更優選為選自由以下述化學式(6)~(8)表示的化合物組成的組的至少1種的化合物。
【化9】
m1cu2o2…(6)cum2o2…(7)cum3o…(8)
上述化學式(6)~(8)中,m1為第2族元素,m2為第13族元素,m3為第5族元素。
作為由化學式(6)表示的化合物的例子,可列舉becu2o2、mgcu2o2、cacu2o2、srcu2o2、bacu2o2、racu2o2等。其中,優選為srcu2o2或bacu2o2。
作為由化學式(7)表示的化合物的例子,可列舉cubo2、cualo2、cugao2、cuino2、cutlo2等。其中,優選為cualo2或cuino2。
作為由化學式(8)表示的化合物的例子,可列舉cuvo、cutao、cunbo等。其中,優選為cutao或cunbo。
即,本發明的p型半導體微粒優選選自由srcu2o2、bacu2o2、cualo2、cuino2、cutao和cunbo組成的組。
p型半導體微粒可以單獨地使用1種,也可將2種以上混合使用。
就p型半導體微粒的數均一次粒徑而言,作為表面處理前的值,優選1~1000nm,更優選10~500nm,進一步優選10~100nm,特別優選10~50nm。如果為1nm以上,能夠充分地發揮p型半導體的性能,粉碎、分級容易,實用性優異。另一方面,如果為1000nm以下,則分散性、涂布性良好,作為感光體的性能優異。
就p型半導體微粒的數均一次粒徑而言,可以通過掃描型電子顯微鏡(日本電子株式會社制造、jsm-7500f)拍攝10萬倍的放大照片、用自動圖像處理解析裝置(株式會社ニレコ制造、“luzexap”軟件1.32版)對通過掃描儀隨機地收進了300個粒子的照片圖像(不包括凝聚粒子)進行解析,由此算出。
作為本發明涉及的p型半導體微粒的制備方法,可列舉激光燒蝕法、燒結法、結晶生長法、脈沖激光蒸鍍法等,但并不限于此。作為具體的制造方法的例子,可列舉日本特開2015-141269號公報、日本特開2007-169089號公報、日本特開2002-114515號公報、kawazoe等,nature,389,939(1997)中公開的方法等。
就固化性組合物中的p型半導體微粒的含量而言,相對于后述的聚合性化合物100質量份,優選10~500質量份,更優選10~100質量份。在10質量份以上的情況下,保護層的空穴傳輸能力良好。另一方面,在500質量份以下的情況下,保護層形成用涂布液的涂布性良好。就其優選的含量而言,與保護層中的p型半導體微粒的優選的含量同等。
從提高保護層的耐磨損性的觀點出發,就本發明涉及的p型半導體微粒而言,優選在表面具有反應性有機基團。通過用具有反應性有機基團的表面處理劑對p型半導體微粒進行處理,能夠將反應性有機基團導入該p型半導體微粒表面。通過進行這樣的導入,p型半導體微粒與后述的聚合性化合物一起發生固化反應,可形成共價鍵。
[表面處理劑]
作為具有反應性有機基團的表面處理劑,使用具有與在p型半導體微粒的表面存在的羥基等的反應性的表面處理劑。作為這樣的表面處理劑,可列舉硅烷偶聯劑、鈦偶聯劑等。作為反應性有機基團,優選環氧基、氧雜環丁烷基等的離子聚合性官能團或自由基聚合性官能團,更優選自由基聚合性官能團。自由基聚合性官能團也與具有聚合性不飽和基團的聚合性化合物反應,能夠形成牢固的保護層。作為自由基聚合性官能團,可列舉乙烯基、丙烯酰基、甲基丙烯酰基等的烯屬不飽和基團,作為表面處理劑,優選具有這些自由基聚合性官能團的硅烷偶聯劑。作為上述這樣的表面處理劑,例如可列舉由下述化學式s-1~s-36表示的化合物等。
【化10】
s-1:ch2=chsi(ch3)(och3)2
s-2:ch2=chsi(och3)3
s-3:ch2=chsicl3
s-4:ch2=chcoo(ch2)2si(ch3)(och3)2
s-5:ch2=chcoo(ch2)2si(och3)3
s-6:ch2=chcoo(ch2)2si(oc2h5)(och3)2
s-7:ch2=chcoo(ch2)3si(och3)3
s-8:ch2=chcoo(ch2)2si(ch3)cl2
s-9:ch2=chcoo(ch2)2sicl3
s-10:ch2=chcoo(ch2)3si(ch3)cl2
s-11:ch2=chcoo(ch2)3sici3
s-12:ch2=c(ch3)coo(ch2)2si(ch3)(och3)2
s-13:ch2=c(ch3)coo(ch2)2si(och3)3
s-14:ch2=c(ch3)coo(ch2)3si(ch3)(och3)2
s-15:ch2=c(ch3)coo(ch2)3si(och3)3
s-16:ch2=c(ch3)coo(ch2)2si(ch3)cl2
s-17:ch2=c(ch3)coo(ch2)2sicl3
s-18:ch2=c(ch3)coo(ch2)3si(ch3)cl2
s-19:ch2=c(ch3)coo(ch2)3sicl3
s-20:ch2=chsi(c2h5)(och3)2
s-21:ch2=c(ch3)si(och3)3
s-22:ch2=c(ch3)si(oc2h5)3
s-23:ch2=chsi(och3)3
s-24:ch2=c(ch3)si(ch3)(och3)2
s-25:ch2=chsi(ch3)cl2
s-26:ch2=chcoosi(och3)3
s-27:ch2=chcoosi(oc2h5)3
s-28:ch2=c(ch3)coosi(och3)3
s-29:ch2=c(ch3)coosi(oc2h5)3
s-30:ch2=c(ch3)coo(ch2)3si(oc2h5)3
s-31:ch2=chcoo(ch2)2si(ch3)2(och3)
s-32:ch2=chcoo(ch2)2si(ch3)(ococh3)2
s-33:ch2=chcoo(ch2)2si(ch3)(onhch3)2
s-34:ch2=chcoo(ch2)2si(ch3)(oc6h5)2
s-35:ch2=chcoo(ch2)2si(c10h21)(och3)2
s-36:ch2=chcoo(ch2)2si(ch2c6h5)(och3)2
其中,優選作為在一個末端具有甲氧基、在另一末端具有甲基丙烯酰基或丙烯酰基的化合物的s-4~s-7及s-12~s-15,從實現本發明的效果的觀點以及成本及可得到性的觀點出發,特別優選s-7或s-15。
另外,作為表面處理劑,在上述的s-1~s-36以外,也可使用具有自由基聚合性官能團的硅烷化合物。這些表面處理劑可以單獨使用或者將2種以上混合使用。
[p型半導體微粒的表面處理方法]
作為p型半導體微粒的表面處理方法,并無特別限制,可列舉如下的方法:制備含有p型半導體微粒、表面處理劑及溶劑的漿料后,使用濕式介質分散型裝置進行了濕式粉碎及表面處理后,將溶劑除去。
所謂作為本發明中使用的表面處理裝置的濕式介質分散型裝置,為具有下述工序的裝置:通過在容器內填充珠粒作為介質、進而使與旋轉軸垂直地安裝了的攪拌圓盤高速旋轉,將凝聚的p型半導體微粒破碎而進行粉碎·分散。作為其構成,只要是對于p型半導體微粒進行表面處理時能夠使p型半導體微粒充分地分散、且進行表面處理的形式,則沒有問題,例如能夠采用立式·臥式型、連續式·間歇式等的各種的樣式。具體地,能夠使用砂磨機、ウルトラビスコミル、珠磨機(パールミル)、谷物磨機(グレンミル)、ダイノミル、攪拌磨(アジテータミル)、動態磨機(ダイナミックミル)等。這些分散型裝置使用球粒、珠粒等的粉碎介質(media)通過沖擊壓壞、摩擦、剪切、剪應力(ズリ応力)等來進行微粉碎、分散。
作為上述濕式介質分散型裝置中使用的珠粒,可以使用以玻璃、氧化鋁、鋯石、氧化鋯、鋼、燧石等作為原材料的球粒,特別優選氧化鋯制、鋯石制的球粒。另外,作為珠粒的大小,通常使用直徑1~2mm左右的珠粒,但在本發明中,優選使用0.1~1.0mm左右的珠粒。
作為濕式介質分散型裝置中使用的圓盤、容器內壁,能夠使用不銹鋼制、尼龍制、陶瓷制等各種的原料的圓盤、容器內壁,但在本發明中,特別優選氧化鋯或碳化硅這樣的陶瓷制的圓盤、容器內壁。
就進行表面處理時的表面處理劑的添加量而言,相對于p型半導體微粒100質量份,優選為0.1~200質量份,更優選為7~70質量份。如果為0.1質量份以上,則p型半導體微粒的分散性優異,表面層的性能良好。另一方面,如果為200質量份以下,則難以發生殘留的表面處理劑引起的感光體的電特性的降低。
就制備上述漿料時的溶劑的添加量而言,相對于p型半導體微粒100質量份,優選30~2000質量份。作為使用的溶劑,例如可列舉甲苯、二甲苯、二氯甲烷、1,2-二氯乙烷、甲乙酮、環己烷、醋酸乙酯、醋酸叔丁酯、甲醇、乙醇、正丙醇、異丙醇、正丁醇、叔丁醇、仲丁醇、甲基溶纖劑、4-甲氧基-4-甲基-2-戊酮、乙基溶纖劑、四氫呋喃、1-二噁烷、1,3-二氧戊環、吡啶、二乙胺等。上述的溶劑可以單獨使用或者將2種以上混合使用。
進行表面處理時的處理溫度優選20~100℃,另外處理時間優選1~24小時。
《n型有機半導體》
n型有機半導體是指使用自由電子作為傳輸電荷的載流子的有機半導體。
本發明的保護層中所含的n型有機半導體優選為選自由以下述通式(1)、(2a)、(2b)、(3a)、(3b)、(4)和(5)表示的化合物組成的組的至少1種的化合物。
[由通式(1)表示的化合物]
【化11】
上述通式(1)中,xa為由下述的化學式(1-1)~(1-5)的任一者表示的4價的基團,從合成的容易性的觀點出發,優選為由苯或萘衍生的4價的基團(由下述化學式(1-1)或(1-3)表示的基團)。
【化12】
xa可以不具有取代基,或者可具有選自由取代或未取代的c1~c8烷基、取代或未取代的c1~c8烷氧基、鹵素原子(f、cl、br或i原子)、取代或未取代的c6~c18芳基、氰基、硝基及羥基組成的組中的至少1個取代基。
作為未取代的c1~c8烷基的例子,可列舉甲基、乙基、正丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、正戊基、正己基、正庚基、正辛基、2-乙基己基等的直鏈狀或分支狀的烷基。
作為未取代的c1~c8烷氧基的例子,可列舉甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、仲丁氧基等。
作為未取代的c6~c18芳基的例子,可列舉苯基、萘基、聯苯基、芴基、蒽基、芘基、薁基、苊基、三聯苯基、菲基等。
上述通式(1)中,r11及r12各自獨立地選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子(f、cl、br或i原子)、取代或未取代的c6~c12芳基、氰基及硝基組成的組。予以說明,取代基的定義與上述的定義相同。r11及r12可以為相同的取代基,也可為不同的取代基。
作為未取代的c3~c12環烷基的例子,可列舉環丙基、環戊基、環己基、金剛烷基等。
作為r11及r12的更優選的具體例,并無特別限制,但可列舉2-三氟甲基苯基、1,2-二甲基丙基、2,6-二甲基苯基、4-戊氧基(ペントキシ)羰基環己基、1-己基庚基等。
由通式(1)表示的化合物可使用市售品、合成品的任一種。在合成品的情況下,通過在溶劑(優選地二甲基甲酰胺等的非質子性極性溶劑)存在下或非存在下使四羧酸二酐與氨基化合物反應,能夠得到所期望的n型有機半導體。作為合成方法的具體例,可列舉日本特開2001-265031號公報、j.am.chem.soc,120,323(1998)、journaloforganicchemistry,72,7287-7293(2007)等中記載的合成方法。
由通式(1)表示的化合物可以單獨地使用1種,也可將2種以上組合使用。予以說明,由通式(1)表示的化合物可以進一步被其他的取代基取代。
作為由通式(1)表示的化合物的例子,并無特別限制,優選下述etm01~06。
【化13】
[由通式(2a)或(2b)表示的化合物]
【化14】
通式(2a)中,xb為取代或未取代的c6~c18亞芳基,作為例子,可列舉亞苯基、亞聯苯基、亞三聯苯基、亞萘基等。其中,xb優選為亞聯苯基。
通式(2a)中,a1及a2各自獨立地為亞噁二唑基。作為亞噁二唑基,可以是1,2,3-亞噁二唑基、1,2,4-亞噁二唑基、1,2,5-亞噁二唑基或1,3,4-亞噁二唑基的任一者,但優選y1及y2中至少一者為1,3,4-亞噁二唑基,更優選y1及y2都為1,3,4-亞噁二唑基。通式(2b)中,a1與通式(2a)中的a1的定義同義。
通式(2a)及(2b)中,優選r21及r22各自獨立地為取代或未取代的c6~c18芳基,更優選為取代或未取代的苯基。另外,在r21及r22中可存在的取代基優選選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組,更優選為取代或未取代的c1~c8烷氧基,進一步更優選為取代或未取代的乙氧基。
未取代的c1~c8烷氧基羰基(-coor)為羧基(-cooh)的氫原子被取代為烷基的基團,作為例子,可列舉甲氧基羰基、乙氧基羰基、正丙氧基羰基、正丁氧基羰基等。
由通式(2a)及(2b)表示的化合物可以使用市售品、合成品的任一種。對于由通式(2a)表示的化合物,在合成品的情況下,可以通過使二羧酰氯(cloc-xb-cocl)與羧酰肼化合物(r-co-nh-nh2)或氨基肟化合物(r-c(nh2)=noh)反應,接著,使生成的羧酸二酰肼或酰氨基肟進行脫水環化反應等而得到。作為合成方法的具體例,可列舉synthesis,1986,#5,第411-413頁,rekkas、j.org.chem.,2009,74(16),第6410-6413頁,mukai等。
由通式(2a)或(2b)表示的化合物可以單獨地使用1種,也可將2種以上組合使用。予以說明,由通式(2a)或(2b)表示的化合物可進一步被其他的取代基取代。
作為由通式(2a)表示的化合物的例子,并無特別限制,優選下述etm201。
【化15】
作為由通式(2b)表示的化合物的例子,并無特別限制,優選將苯基取代了的或者未取代的2,5-二苯基-1,3,4-噁二唑,作為具體例,可列舉下述etm202、etm203等。
【化16】
[由通式(3a)表示的化合物]
【化17】
通式(3a)中,r301~r304各自獨立地為選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團,可相互連接而形成環結構。此時,上述環結構可以為芳香環或非芳香環的任一種(優選為芳香環),可具有至少1個選自由硫(s)、氮(n)和氧(o)組成的組的雜原子。
由通式(3a)表示的化合物可以使用市售品、合成品的任一種。另外,由通式(3a)表示的化合物可以單獨地使用1種,也可將2種以上組合使用。
作為由通式(3a)表示的化合物的例子,并無特別限制,可列舉2,5-二-叔丁基-對-苯醌、2,6-二-叔丁基-對-苯醌、2,3,5,6-四-叔丁基-對-苯醌、萘醌、蒽醌、下述etm301~etm303等。
【化18】
[由通式(3b)表示的化合物]
【化19】
通式(3b)中,r305~r312各自獨立地為選自由氫原子、取代或未取代的c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、鹵素原子、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團,可相互連接而形成環結構。此時,上述環結構可以是芳香環或非芳香環的任一種(優選為芳香環),可具有至少1個選自由硫(s)、氮(n)和氧(o)組成的組的雜原子。其中,r305~r312優選各自獨立地為氫原子、取代或未取代的c1~c4烷基,更優選為氫原子或叔丁基。進而,r305、r308、r309及r312中,優選地1個、更優選地2個、進一步更優選地3個、特別優選地4個優選被氫原子以外的取代基取代。
作為由通式(3b)表示的化合物的例子,并無特別限制,可列舉3,3’,5,5’-四-叔丁基-4,4’-聯苯醌(etm311)、3,3’,5,5’-四甲基-4,4’-聯苯醌、3,3’,5,5’-四乙基-4,4’-聯苯醌、3,3’,5,5’-四-正丁基-4,4’-聯苯醌、3,3’-二-叔丁基-5,5’-二甲基-4,4’-聯苯醌(etm312)、聯蒽酮(etm313)、下述etm314等,優選3,3’,5,5’-四-叔丁基-4,4’-聯苯醌(etm311)。
【化20】
由通式(3b)表示的化合物可使用市售品、合成品的任一種。作為市售品,可以從sigma-aldrich公司、東京化成工業株式會社等購入。
由通式(3b)表示的化合物可以單獨地使用1種,也可將2種以上組合使用。予以說明,由通式(3b)表示的化合物可以進一步被其他取代基取代。
[由通式(4)表示的化合物]
【化21】
通式(4)中,q1~q6各自獨立地為碳原子或氮原子,優選地,q1~q6中的至少1個為氮原子,更優選地,q1或q4為氮原子,進一步更優選地,q1為氮原子。
另外,通式(4)中,r41及r42各自獨立地為選自由氫原子、c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、取代或未取代的c6~c18芳基、氰基、硝基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團,r41及r42可相互連接而形成環結構。其中,r41及r42中的至少一者優選為取代或未取代的c6~c18芳基,更優選為取代或未取代的苯基。
通式(4)中,r43及r44各自獨立地為選自由氫原子、c1~c8烷基、取代或未取代的c3~c12環烷基、取代或未取代的c1~c8烷氧基、取代或未取代的c6~c18芳基、氰基、硝基、取代或未取代的c2~c24雜芳基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團。
作為未取代的c2~c24雜芳基的例子,可列舉呋喃基、噻吩基、吡啶基、噠嗪基、嘧啶基、吡嗪基、三嗪基、咪唑基、吡唑基、噻唑基、喹唑啉基、咔唑基、咔啉基、氮雜咔唑基、苯并呋喃基、二苯并呋喃基、氮雜二苯并呋喃基、苯并噻吩基、吲哚基、二苯并噻吩基、苯并咪唑基、噁唑基、苯并噁唑基、喹喔啉基、異噁唑基、異噻唑基、吲哚并咔唑基、六氮雜苯并[9,10]菲基、苯并二呋喃基、苯并二噻吩基、磷酰基(ホスフォル基)、甲硅烷基(シロリル基)、(甲)硼烷基(ボリル基)、聯吡啶基等。
其中,r43及r44中的至少一者優選為取代或未取代的c2~c24雜芳基,更優選為取代或未取代的c2~c24含氮雜芳基,進一步更優選為取代或未取代的c5~c12含氮雜芳基,特別優選為咔唑基。
由通式(4)表示的化合物可使用市售品、合成品的任一種。作為合成方法的具體例,可列舉science,1998,282,913~915、j.org.chem.,2002,vol.67,9392~9396、j.org.chem.,2006,vol.71,7826~7834、j.am.chem.soc.,vol.124,no.1,2002,49~57、國際公開第2006/002731號小冊子、j.c.s.,[section]b,pysicalorganic1996,733-735、chem.lett.,1982,1195~1198等。
由通式(4)表示的化合物可單獨地使用1種,也可將2種以上組合使用。予以說明,由通式(4)表示的化合物可以進一步被其他的取代基取代。
作為由通式(4)表示的化合物的例子,并無特別限制,優選下述etm401。
【化22】
[由通式(5)表示的化合物]
【化23】
通式(5)中,z為碳原子或氮原子,優選為碳原子。
另外,通式(5)中,r51及r52的至少一者為腈基或者取代或未取代的c1~c8烷氧基羰基,優選為腈基,更優選地,r51及r52都為腈基。
另外,通式(5)中,r53~r60各自獨立地為選自由氫原子、c1~c8烷基、取代或未取代的c6~c18芳基、羧基及取代或未取代的c1~c8烷氧基羰基組成的組的基團。
r57~r60中,優選地1個、更優選地2個、進一步更優選地3個、特別優選地4個全部為氫原子。
r53~r56中,至少1個優選為取代或未取代的c6~c18芳基,優選為取代或未取代的苯基。另外,r53~r56中,剩余的基團優選為取代或未取代的c1~c8烷氧基羰基,更優選為取代或未取代的c1~c4烷氧基羰基,進一步更優選為取代或未取代的甲氧基羰基(-cooch3)。
由通式(5)表示的化合物可以使用市售品、合成品的任一種。另外,由通式(5)表示的化合物可以單獨地使用1種,也可將2種以上組合使用。予以說明,由通式(5)表示的化合物可以進一步被取代。
作為由通式(5)表示的化合物的例子,并無特別限制,優選下述etm501。
【化24】
由上述(1)、(2a)、(2b)、(3a)、(3b)、(4)及(5)表示的化合物可以單獨地使用1種,也可將2種以上組合使用。
就固化性組合物中的n型有機半導體的含量而言,相對于后述的聚合性化合物100質量份,優選0.1~50質量份,更優選0.5~20質量份,進一步優選1.0~15質量份,特別優選5.0~10質量份。如果為0.1質量份以上,則良好地發揮作為n型有機半導體的功能。另一方面,如果為50質量份以下,則保護層的耐久性優異,能夠實現感光體的高壽命化。其優選的含量與保護層中的n型有機半導體的優選的含量同等。
另外,固化性組合物中的p型半導體微粒及n型有機半導體的含有比例優選1:1~50:1,更優選5:1~25:1,進一步更優選10:1~20:1。通過在上述范圍內,耐圖像記憶性優異,且能夠良好地抑制翳影的發生。其優選的含有比例與保護層中的優選的含有比例同等。
《聚合性化合物》
作為本發明涉及的保護層中可使用的聚合性化合物,通過紫外線、電子束等的活性能量線照射而聚合(固化),形成聚苯乙烯、聚(甲基)丙烯酸酯等、一般作為感光體的粘結劑樹脂所使用的樹脂的單體是優選的。特別優選苯乙烯系單體、丙烯酸系單體、甲基丙烯酸系單體、乙烯基甲苯系單體、醋酸乙烯酯系單體、n-乙烯基吡咯烷酮系單體。
其中,從用少的光量或短的時間就可以固化的角度出發,優選具有丙烯酰基(ch2=chco-)或甲基丙烯酰基(ch2=cch3co-)等的聚合性不飽和基團的自由基聚合性單體或它們的低聚物。即,本發明涉及的聚合性化合物優選具有(甲基)丙烯酰基。
本發明中,上述的聚合性化合物可以單獨地使用,也可將2種以上混合使用。另外,這些聚合性化合物可以使用單體,也可低聚物化而使用。
作為這些聚合性化合物,例如可以例示以下的化合物,但并不限定于這些。另外,以下例示的聚合性化合物為公知的,可以作為市售品得到。
【化25】
上述式中,r表示丙烯酰基,r’表示甲基丙烯酰基(參照下述化學式)。
【化26】
上述中,作為本發明中優選使用的聚合性化合物,可列舉上述例示化合物m1、上述例示化合物m2、上述例示化合物m3。
上述聚合性化合物優選使用具有3個以上聚合性不飽和基團的化合物。另外,聚合性化合物可以將2種以上的化合物并用,即使是這種情況下,相對于聚合性化合物100質量%,優選使用50質量%以上的具有3個以上聚合性不飽和基團的化合物。另外,就聚合性不飽和基團當量、即“具有聚合性不飽和基團的化合物的分子量/不飽和基團數”而言,優選1000以下,更優選500以下。由此,保護層的交聯密度升高,耐磨損性提高。
[聚合引發劑]
本發明中,通過使固化性組合物固化而形成保護層,在使固化性組合物固化時,可使用通過電子束開裂而使其反應的方法;添加自由基聚合引發劑而用光、熱來使其反應的方法等。就聚合引發劑而言,光聚合引發劑、熱聚合引發劑都可以使用,也可以將光聚合引發劑及熱聚合引發劑這兩者并用。
作為熱聚合引發劑,例如可列舉2,2’-偶氮二異丁腈、2,2’-偶氮二(2,4-二甲基偶氮二戊腈)、2,2’-偶氮二(2-甲基丁腈)等的偶氮化合物;過氧化苯甲酰(bpo)、二叔丁基過氧化氫、叔丁基過氧化氫、過氧化氯苯甲酰、過氧化二氯苯甲酰、過氧化溴甲基苯甲酰、過氧化月桂酰等的過氧化物等。
作為光聚合引發劑,例如可列舉二乙氧基苯乙酮、2,2-二甲氧基-1,2-二苯基乙烷-1-酮、1-羥基-環己基-苯基-酮、4-(2-羥基乙氧基)苯基-(2-羥基-2-丙基)酮、2-芐基-2-二甲基氨基-1-(4-嗎啉代苯基)丁酮-1(“irgacure(注冊商標)369”:basfジャパン株式會社制造)、2-羥基-2-甲基-1-苯基丙烷-1-酮、2-甲基-2-嗎啉代(4-甲硫基苯基)丙烷-1-酮、1-苯基-1,2-丙二酮-2-(鄰-乙氧基羰基)肟等的苯乙酮系或縮酮系光聚合引發劑;苯偶姻、苯偶姻甲基醚、苯偶姻乙基醚、苯偶姻異丁基醚、苯偶姻異丙基醚等的苯偶姻醚系光聚合引發劑;二苯甲酮、4-羥基二苯甲酮、鄰-苯甲酰基苯甲酸甲酯、2-苯甲酰基萘、4-苯甲酰基聯苯、4-苯甲酰基苯基醚、丙烯酰基化二苯甲酮、1,4-苯甲酰基苯等的二苯甲酮系光聚合引發劑;2-異丙基噻噸酮、2-氯噻噸酮、2,4-二甲基噻噸酮、2,4-二乙基噻噸酮、2,4-二氯噻噸酮等的噻噸酮系光聚合引發劑等。
作為其他的光聚合引發劑,例如可列舉乙基蒽醌、2,4,6-三甲基苯甲酰基二苯基氧化膦、2,4,6-三甲基苯甲酰基苯基乙氧基氧化膦、雙(2,4,6-三甲基苯甲酰基)苯基氧化膦(“irgacure(注冊商標)819”:basfジャパン株式會社制造)、雙(2,4-二甲氧基苯甲酰基)-2,4,4-三甲基戊基氧化膦、甲基苯基乙醛酸酯、9,10-菲、吖啶系化合物、三嗪系化合物、咪唑系化合物等。另外,也可以將具有光聚合促進效果的光聚合促進劑單獨地使用或者與上述光聚合引發劑并用而使用。作為光聚合促進劑,例如可列舉三乙醇胺、甲基二乙醇胺、4-二甲基氨基苯甲酸乙酯、4-二甲基氨基苯甲酸異戊酯、苯甲酸(2-二甲基氨基)乙酯、4,4’-二甲基氨基二苯甲酮等。
作為自由基聚合引發劑,優選光聚合引發劑,其中,優選烷基苯基酮系化合物、或氧化膦系化合物。特別優選具有α-氨基烷基苯基酮結構、或酰基氧化膦結構的化合物。
這些聚合引發劑可以使用1種或者將2種以上混合使用。就固化性組合物中的聚合引發劑的含量而言,相對于聚合性化合物的100質量份,優選0.5~30質量份,更優選2~20質量份,進一步優選5~15質量份。
《其他的添加劑》
在本發明涉及的保護層中,除了上述成分以外,根據需要可含有n型半導體微粒、潤滑劑粒子、流平劑(例如,硅油等)、抗氧化劑等的添加劑。可將該成分添加到本發明涉及的固化性組合物中。即,在本發明涉及的固化物中可含有該成分或/和該成分的反應物。
作為n型半導體微粒,可列舉sno2、tio2、al2o3等。n型半導體微粒可以使用采用氣相法、氯法(塩素法)、硫酸法、等離子體法、電解法等的公知的方法所制備的產物。即使對于n型半導體微粒,優選通過與p型半導體微粒同樣地實施表面處理而在表面具有反應性有機基團。另外,就n型半導體微粒的數均一次粒徑而言,作為表面處理前的值,優選1~1000nm,更優選10~500nm,進一步更優選10~100nm,特別優選10~50nm。予以說明,數均一次粒徑的測定方法與p型半導體微粒同樣。
作為潤滑劑粒子,可使用含有氟原子的樹脂粒子。作為含有氟原子的樹脂粒子,優選選自四氟乙烯樹脂、三氟氯乙烯樹脂、六氟氯乙烯丙烯樹脂、氟乙烯樹脂、偏氟乙烯樹脂、二氟二氯乙烯樹脂、及它們的共聚物中的至少1種,特別優選四氟乙烯樹脂及偏氟乙烯樹脂。
(保護層的制作方法)
本發明涉及的保護層可以通過以下來形成:制作將p型半導體微粒、n型有機半導體、聚合性化合物、及根據需要的添加劑(聚合引發劑、p型半導體微粒以外的無機微粒、潤滑劑粒子等)在溶劑中混合的固化性組合物(保護層形成用涂布液),將該固化性組合物在后述的感光層上涂布后,使其干燥及固化。
在上述涂布、干燥及固化的過程中,聚合性化合物間的反應、p型半導體微粒具有反應性有機基團的情況下聚合性化合物與p型半導體微粒的反應性有機基團之間的反應、以及具有反應性有機基團的p型半導體微粒間的反應等進行,形成保護層。
作為固化性組合物(保護層形成用涂布液)中使用的溶劑,只要能夠使p型半導體微粒、n型有機半導體及聚合性化合物、和根據需要的上述的添加劑溶解或分散,則能夠使用任一溶劑。具體地,例如可列舉甲醇、乙醇、丙醇、異丙醇、1-丁醇、2-丁醇、叔丁醇、芐醇、甲苯、二甲苯、二氯甲烷、甲乙酮、環己烷、醋酸乙酯、醋酸丁酯、甲基溶纖劑、乙基溶纖劑、四氫呋喃、1-二噁烷、1,3-二氧戊環、吡啶、二乙胺等,但并不限定于這些。這些溶劑可以單獨使用或者將2種以上組合使用。
就固化性組合物(保護層形成用涂布液)中的溶劑的含量而言,相對于涂布液全體,優選為10~90質量%,優選為30~80質量%。
對固化性組合物(保護層形成用涂布液)的制造方法也無特別限制,可以將p型半導體微粒子、n型有機半導體、聚合性化合物、及根據需要的上述的添加劑加入溶劑中,進行攪拌混合直至溶解或分散。另外,對溶劑的量也無特別限制,可以適當地調節以使得固化性組合物(保護層形成用涂布液)形成適于涂布作業的粘度。
對涂布方法并無特別限制,例如可以使用浸涂法、噴涂法、旋涂法、珠粒涂布法(ビードコーティング法)、刮刀涂布法、射束涂布法(ビームコーティング法)、滑動料斗法(スライドホッパー法)、圓形滑動料斗法(円形スライドホッパー法)等的公知的方法。
將上述涂布液涂布后,進行自然干燥或熱干燥,形成涂膜后,照射活性能量線進行固化,生成含有作為單體成分的聚合性化合物及被表面處理了的無機微粒的組合物的固化物。作為活性能量線,更優選紫外線、電子束,進一步優選紫外線。
作為紫外線光源,只要是產生紫外線的光源,則可以無限制地使用。例如,可以使用低壓汞燈、中壓汞燈、高壓汞燈、超高壓汞燈、碳弧燈、金屬鹵化物燈、氙燈、閃光(脈沖)氙燈等。照射條件因各個燈而不同,但光的照射強度優選為1000~5000mw/cm2,更優選為3000~4000mw/cm2。另外,涂布膜中的光的照射強度優選為500~3000mw/cm2,更優選為1000~2000mw/cm2。另外,涂膜的固化工藝優選在非活性氣體氣氛下進行,更優選在氮氣氛下進行。
作為用作電子束源的電子束照射裝置,并無特別限制,一般地,作為電子束照射用的電子束加速機,優選使用價格比較便宜且可得到大輸出功率的簾幕射束方式的裝置。電子束照射時的加速電壓優選為100~300kv。作為吸收線量,優選為0.5~10mrad。
作為用于得到必要的活性能量線的照射量的照射時間,優選0.1秒~10分鐘,從作業效率的觀點出發,優選0.1秒~5分鐘。
在形成保護層的過程中,可以在照射活性能量線的前后、照射活性能量線中進行干燥,進行干燥的時機可以將它們組合而適當地選擇。
干燥的條件可以根據溶劑的種類、膜厚等來適當地選擇。干燥溫度優選為20~180℃,更優選為20~100℃,進一步優選為20~50℃。干燥時間優選為1~200分鐘,更優選為5~100分鐘,進一步更優選為10~30分鐘。
保護層的干燥膜厚優選0.1~15μm,更優選1.0~10μm,特別優選2.0~5.0μm。在保護層的膜厚為15μm以下的情況下,保護層的膜厚不均少,所形成的圖像的清晰性良好。另一方面,在保護層的膜厚為0.1μm以上的情況下,可以提高感光體的壽命及耐磨損性。
[導電性支承體]
就本發明中使用的導電性支承體而言,只要具有導電性,則可以是任何導電性支承體,例如可列舉將鋁、銅、鉻、鎳、鋅及不銹鋼等的金屬成型為鼓或片狀的產物、將鋁、銅等的金屬箔層合于塑料膜的產物;將鋁、氧化銦及氧化錫等蒸鍍于塑料膜的產物、將導電性物質單獨地或與粘結劑樹脂一起涂布而設置了導電層的金屬、塑料膜及紙等。
[感光層]
本發明涉及的感光層可以是將電荷產生功能和電荷傳輸功能賦予了1個層的單層結構,更優選使這些功能分離到電荷產生層和電荷傳輸層的多層結構。通過形成為這樣的結構,可以抑制與反復使用相伴的殘留電位的上升。在負帶電性感光體的情況下,在電荷產生層上層疊有電荷傳輸層。在正帶電性感光體的情況下,在電荷傳輸層上層疊有電荷產生層。
以下對電荷產生層及電荷傳輸層分別進行說明。
(電荷產生層)
本發明的感光體中使用的電荷產生層優選含有電荷產生物質和粘結劑樹脂,優選使電荷產生物質在粘結劑樹脂溶液中分散、涂布、干燥而形成。
作為電荷產生物質,例如可列舉蘇丹紅和ダイアン藍等的偶氮顏料、芘醌及蒽嵌蒽醌等的醌顏料、喹菁(キノシアニン)顏料、苝顏料、靛藍及硫靛等的靛藍顏料、皮蒽酮及二鄰苯二甲酰基芘等的多環醌顏料、酞菁顏料等,但并不限定于這些。優選為多環醌顏料和氧鈦酞菁顏料。這些電荷產生物質可以單獨地使用,或者以在公知的粘結劑樹脂中分散的形態使用。
作為電荷產生層的粘結劑樹脂,能夠使用公知的樹脂,例如可列舉聚苯乙烯樹脂、聚乙烯樹脂、聚丙烯樹脂、丙烯酸系樹脂、甲基丙烯酸系樹脂、氯乙烯樹脂、醋酸乙烯酯樹脂、聚乙烯醇縮丁醛樹脂、環氧樹脂、聚氨酯樹脂、酚醛樹脂、聚酯樹脂、醇酸樹脂、聚碳酸酯樹脂、有機硅樹脂、蜜胺樹脂、以及包含這些樹脂中的2個以上的共聚物樹脂(例如,氯乙烯-醋酸乙烯酯共聚物樹脂、氯乙烯-醋酸乙烯酯-馬來酸酐共聚物樹脂)和聚乙烯基咔唑樹脂等,但并不限定于這些。優選為聚乙烯醇縮丁醛樹脂。
就電荷產生層而言,優選使用分散機將電荷產生物質分散在用溶劑將粘結劑樹脂溶解了的溶液中而制備涂布液、用涂布機將涂布液涂布為一定的膜厚、將涂布膜干燥而形成。作為涂布方法,可以使用與上述的保護層同樣的方法。
作為用于將電荷產生層中使用的粘結劑樹脂溶解、涂布的溶劑,例如可列舉甲苯、二甲苯、二氯甲烷、1,2-二氯乙烷、甲乙酮、環己烷、醋酸乙酯、醋酸叔丁酯、甲醇、乙醇、丙醇、丁醇、甲基溶纖劑、4-甲氧基-4-甲基-2-戊酮、乙基溶纖劑、四氫呋喃、1-二噁烷、1,3-二氧戊環、環己酮、吡啶和二乙胺等,但并不限定于這些。這些溶劑可以單獨使用或者將2種以上混合使用。
作為電荷產生物質的分散手段,可以使用超聲波分散機、球磨機、砂磨機及均相混合機等,但并不限定于這些。
就相對于粘結劑樹脂的電荷產生物質的混合比例而言,相對于粘結劑樹脂100質量份,優選1~600質量份,更優選50~500質量份。
電荷產生層的干燥膜厚因電荷產生物質的特性、粘結劑樹脂的特性及混合比例等而不同,優選為0.01~5μm,更優選為0.05~3μm。予以說明,通過在將電荷產生層用的涂布液在涂布前將異物、凝聚物過濾,能夠防止圖像缺陷的產生。予以說明,電荷產生層也可以通過將上述的顏料真空蒸鍍而形成。
(電荷傳輸層)
本發明的感光體中使用的電荷傳輸層優選含有電荷傳輸物質和粘結劑樹脂,優選使電荷傳輸物質在粘結劑樹脂溶液中溶解、涂布、干燥而形成。
作為電荷傳輸物質,作為傳輸電荷(空穴)的物質,例如可列舉三苯基胺衍生物、腙化合物、苯乙烯基化合物、聯苯胺化合物、丁二烯化合物等。
電荷傳輸層用的粘結劑樹脂可以使用公知的樹脂,可列舉聚碳酸酯樹脂、聚丙烯酸酯樹脂、聚酯樹脂、聚苯乙烯樹脂、苯乙烯-丙烯腈共聚物樹脂、聚甲基丙烯酸酯樹脂及苯乙烯-甲基丙烯酸酯共聚物樹脂等,優選聚碳酸酯樹脂。這些粘結劑樹脂可以單獨地使用或者將2種以上混合使用。進而,從耐開裂性、耐磨損性、帶電特性等的觀點出發,優選雙酚a(bpa)、雙酚z(bpz)、二甲基bpa、bpa-二甲基bpa共聚物等。
電荷傳輸層優選通過溶解粘結劑樹脂和電荷傳輸物質而制備涂布液、將涂布液用涂布機涂布到一定的膜厚、將涂布膜干燥而形成。作為涂布方法,可以使用與后述的保護層同樣的方法。
作為用于溶解上述粘結劑樹脂和電荷傳輸物質的溶劑,例如可列舉甲苯、二甲苯、二氯甲烷、1,2-二氯乙烷、甲乙酮、環己酮、醋酸乙酯、醋酸丁酯、甲醇、乙醇、丙醇、丁醇、四氫呋喃、1,4-二噁烷、1,3-二氧戊環、吡啶及二乙胺等,但并不限定于這些。這些溶劑可以單獨地使用或者將2種以上混合使用。
就相對于粘結劑樹脂的電荷傳輸物質的混合比例而言,相對于粘結劑樹脂100質量份,優選10~500質量份,更優選20~250質量份。
電荷傳輸層的干燥膜厚因電荷傳輸物質的特性、粘結劑樹脂的特性及混合比例等而不同,但優選為5~40μm,更優選為10~30μm。
在電荷傳輸層中可添加抗氧化劑、電子導電劑、穩定劑、硅油等。作為抗氧化劑的例子,例如可列舉日本特開2000-305291號公報中記載的化合物。作為電子導電劑的例子,例如可列舉日本特開昭50-137543號公報、日本特開昭58-76483號公報等中記載的化合物。
[其他層]
在本發明中,在導電性支承體上可具有感光層及保護層以外的層。特別地,從防故障等的觀點出發,優選在導電性支承體與感光層之間設置具有阻隔功能和粘接功能的中間層。
中間層可以通過將酪蛋白、聚乙烯醇、硝基纖維素、乙烯-丙烯酸共聚物、聚酰胺樹脂、聚氨酯樹脂、或明膠等的粘結劑樹脂溶解于公知的溶劑、制作涂布液后,通過浸涂法等的與上述保護層同樣的涂布方法及干燥方法來形成。其中優選可溶于醇的聚酰胺樹脂。這些粘結劑樹脂可以單獨地使用或者將2種以上組合使用。
另外,為了調節中間層的電阻,可以含有各種的導電性粒子、金屬氧化物粒子等的無機粒子。例如,可以使用氧化鋁、氧化鋅、氧化鈦、氧化錫、氧化銻、氧化銦、氧化鉍等的各種金屬氧化物粒子、錫摻雜的氧化銦(ito)、銻摻雜的氧化錫(ato)、氧化鋯等的粒子。
這些無機粒子可以單獨使用或者將2種以上組合使用。在將2種以上混合的情況下,可采取固溶體或熔合的形態。
無機粒子的平均一次粒徑優選為300nm以下,更優選為100nm以下,進一步優選為50nm以下。另一方面,對平均一次粒徑的下限值并無特別限制,優選為10nm以上,更優選為20nm以上。無機粒子的平均一次粒徑可以用與p型半導體微粒同樣的方法來測定。
作為在中間層的涂布液中所使用的溶劑,優選良好地分散上述那樣的金屬氧化物粒子、將粘結劑樹脂、特別是聚酰胺樹脂溶解的溶劑。具體地,甲醇、乙醇、正丙醇、異丙醇、正丁醇、叔丁醇、仲丁醇等的碳數1~4的醇類由于聚酰胺樹脂的溶解性和涂布性能優異,因此優選。這些溶劑可以單獨地使用或者將2種以上組合使用。另外,為了提高保存性及無機粒子的分散性,可以將上述的溶劑與助溶劑并用。作為得到優選的效果的助溶劑,可列舉芐醇、甲苯、二氯甲烷、環己酮、四氫呋喃等。
對形成中間層的方法并無特別限制,使粘結劑樹脂溶解于上述的溶劑中,接著使用超聲波分散機、珠磨機、球磨機、砂磨機、或均相混合機等的裝置使無機粒子分散,制備涂布液后,將該涂布液在導電性支承體上涂布為所期望的厚度。然后,使涂布的層干燥,完成中間層。中間層的干燥方法可以根據溶劑的種類、膜厚適當地選擇,但優選加熱干燥。
用于中間層形成的涂布液中的粘結劑樹脂的濃度根據中間層的膜厚、生產速度適當地選擇。
在使無機粒子分散時,相對于粘結劑樹脂的無機粒子的混合比例,相對于粘結劑樹脂100質量份,優選20~400質量份,更優選50~350質量份。
中間層的干燥膜厚優選0.1~15μm,更優選0.3~10μm。
<圖像形成裝置>
本發明也提供具備上述的電子照相感光體的圖像形成裝置。
本發明涉及的圖像形成裝置具有:(1)至少具有本發明的保護層的電子照相感光體、(2)對上述的電子照相感光體表面進行帶電的帶電手段、(3)對通過帶電手段而帶電了的電子照相感光體表面進行像曝光而進行潛像形成的曝光手段、(4)使通過曝光手段所形成了的潛像顯像化而形成調色劑圖像的顯影手段、(5)將通過顯影手段在電子照相感光體表面形成了的調色劑圖像轉印至用紙等的轉印介質或轉印帶上的轉印手段。
予以說明,在使電子照相感光體帶電的帶電手段中,優選使用非接觸帶電裝置。作為非接觸帶電裝置,可以列舉電暈帶電裝置、電暈管帶電裝置、高壓艙帶電裝置(スコロトロン帯電裝置)。
圖2為對表示本發明的實施方式之一的彩色圖像形成裝置的一例進行說明的剖面構成圖。
該彩色圖像形成裝置稱為串聯型彩色圖像形成裝置,由4組的圖像形成單元10y、10m、10c、10bk、環形帶狀中間轉印體單元70、給紙輸送手段21及定影手段24構成。在圖像形成裝置的主體a的上部配置有原稿圖像讀取裝置sc。
形成黃色的圖像的圖像形成單元10y具有:在作為第1像負載體的鼓狀的感光體1y的周圍配置的帶電手段(帶電工序)2y、曝光手段(曝光工序)3y、顯影手段(顯影工序)4y、作為一次轉印手段(一次轉印工序)的一次轉印輥5y、清潔手段6y。形成品紅色的圖像的圖像形成單元10m具有:作為第1像負載體的鼓狀的感光體1m、帶電手段2m、曝光手段3m、顯影手段4m、作為一次轉印手段的一次轉印輥5m、清潔手段6m。形成青色的圖像的圖像形成單元10c具有:作為第1像負載體的鼓狀的感光體1c、帶電手段2c、曝光手段3c、顯影手段4c、作為一次轉印手段的一次轉印輥5c、清潔手段6c。形成黑色圖像的圖像形成單元10bk具有:作為第1像負載體的鼓狀的感光體1bk、帶電手段2bk、曝光手段3bk、顯影手段4bk、作為一次轉印手段的一次轉印輥5bk、清潔手段6bk。
上述4組的圖像形成單元10y、10m、10c、10bk以感光體鼓1y、1m、1c、1bk為中心,由帶電手段2y、2m、2c、2bk、曝光手段3y、3m、3c、3bk、顯影手段4y、4m、4c、4bk、及對感光體鼓1y、1m、1c、1bk進行清潔的清潔手段6y、6m、6c、6bk構成。
上述圖像形成單元10y、10m、10c、10bk只是在感光體1y、1m、1c、1bk各自形成的調色劑圖像的顏色不同,為相同的構成,以圖像形成單元10y為例詳細地說明。
就圖像形成單元10y而言,在作為像形成體的感光體鼓1y的周圍配置帶電手段2y(以下簡稱為帶電手段2y、或者、帶電器2y)、曝光手段3y、顯影手段4y、清潔手段6y(以下簡稱為清潔手段6y、或者、清潔刮刀6y),在感光體鼓1y上形成黃色(y)的調色劑圖像。另外,在本實施方式中,設置成:在該圖像形成單元10y中至少感光體鼓1y、帶電手段2y、顯影手段4y、清潔手段6y一體化。
帶電手段2y為對于感光體鼓1y給予一樣的電位的手段,本實施方式中,將電暈放電型的帶電器2y用于感光體鼓1y。
就曝光手段3y而言,為在通過帶電器2y給予了一樣的電位的感光體鼓1y上基于圖像信號(黃色)進行曝光、形成與黃色的圖像對應的靜電潛像的手段,作為該曝光手段3y,使用由在感光體鼓1y的軸向以陣列狀將發光元件排列的led和成像元件(商品名;セルフォック(注冊商標)透鏡)構成的曝光手段、或者、激光光學系等。
作為本發明的圖像形成裝置,可以將上述的感光體、和顯影器、清潔器等的構成要素作為處理盒(圖像形成單元)一體地結合而構成,使該圖像形成單元對于裝置主體裝卸自由地構成。另外,也可將帶電器、像曝光器、顯影器、轉印或分離器、及清潔器的至少1個與感光體一起一體地支承而形成處理盒(圖像形成單元),形成向裝置主體裝卸自由的單一圖像形成單元,也可使用裝置主體的軌道等引導手段而形成裝卸自由的構成。
環形帶狀中間轉印體單元70被多個輥卷繞,具有作為可轉動地被支承的半導電性環形帶狀的第2像負載體的環形帶狀中間轉印體77。
就由圖像形成單元10y、10m、10c、10bk形成的各色的圖像而言,通過作為一次轉印手段的一次轉印輥5y、5m、5c、5bk,被逐次轉印到進行轉動的環形帶狀中間轉印體77上,形成合成了的彩色圖像。就作為在給紙盒20內所容納的作為轉印材料(對定影了的最終圖像進行負載的支承體:例如普通紙、透明片材等)的轉印材料p而言,通過給紙手段21而被給紙,經由多個中間輥22a、22b、22c、22d和定位輥23,被輸送到作為二次轉印手段的二次轉印輥5b,在轉印材料p上進行二次轉印,一并地轉印彩色圖像。就將彩色圖像轉印了的轉印材料p而言,通過定影手段24被定影處理,由排紙輥25夾持而被載置于機外的排紙托盤26上。其中,將中間轉印體、轉印材料等的感光體上形成了的調色劑圖像的轉印支承體總稱為轉印介質。
另一方面,通過作為二次轉印手段的二次轉印輥5b將彩色圖像轉印于轉印材料p后,就將轉印材料p曲率分離了的環形帶狀中間轉印體77而言,通過清潔手段6b將殘留調色劑除去。
圖像形成處理中,一次轉印輥5bk總是抵接于感光體1bk。其他的一次轉印輥5y、5m、5c只在彩色圖像形成時才抵接于各自對應的感光體1y、1m、1c。
二次轉印輥5b只在轉印材料p通過其而進行二次轉印時才抵接于環形帶狀中間轉印體77。
另外,可以從裝置主體a將殼體80經由支承軌82l、82r來抽出。
殼體80由圖像形成單元10y、10m、10c、10bk、和環形帶狀中間轉印體單元70組成。
將圖像形成單元10y、10m、10c、10bk在垂直方向上縱列配置。在感光體1y、1m、1c、1bk的圖示左側方配置有環形帶狀中間轉印體單元70。環形帶狀中間轉印體單元70由將輥71、72、73、74進行卷繞而可轉動的環形帶狀中間轉印體77、1次轉印輥5y、5m、5c、5bk和清潔手段6b組成。
本發明也提供使用上述的圖像形成裝置來形成電子照相圖像的圖像形成方法。根據本發明的圖像形成方法,能夠長期穩定地形成良好的電子照相圖像。
實施例
使用以下的實施例及比較例對本發明的效果進行說明。不過,本發明的技術范圍并不只限于以下的實施例。應予說明,在下述實施例中,只要沒有特別說明,操作在室溫(20~25℃)下進行。另外,只要沒有特別說明,“%”和“份”分別意味著“質量%”和“質量份”。
<p型半導體微粒的制備>
[制造例1-1]p型半導體微粒1的制備
將al2o3及cu2o以1:1的摩爾比混合,在ar氣氛中在1100℃的溫度下臨時燒結4天后,成型為粒料狀,進而在1100℃下燒結2天,由此得到了燒結體。然后,粗粉碎直至數100μm后,使用得到的粗粒子和溶劑,使用濕式介質分散型裝置進行粉碎,得到數均一次粒徑為20nm的cualo2微粒。予以說明,就數均一次粒徑而言,通過掃描型電子顯微鏡(日本電子株式會社制造、jsm-7500f)拍攝10萬倍的放大照片,用自動圖像處理解析裝置(株式會社ニレコ制造、“luzexap”軟件1.32版)對通過掃描儀隨機地取入了300個粒子的照片圖像(不包括凝聚粒子)進行解析,由此算出。應予說明,對于在以下的制造例中得到的p型半導體微粒也通過同樣的方法測定了數均一次粒徑。
將得到的cualo2微粒100質量份、作為表面處理劑的3-甲基丙烯酰氧基丙基三甲氧基硅烷“kbm-503”(信越化學工業株式會社制造)30質量份、及甲乙酮1000質量份裝入濕式砂磨機(直徑0.5mm的氧化鋁珠粒),在30℃下混合6小時,然后,將甲乙酮和氧化鋁珠粒過濾分離,在60℃下干燥,制備了p型半導體微粒1。
[制造例1-2]p型半導體微粒2的制備
將in2o3及cu2o以1:1的摩爾比混合,在ar氣氛中在1100℃的溫度下臨時燒結4天后,成型為粒料狀,進而在1100℃燒結2天,由此得到了燒結體。然后,粗粉碎直至數100μm后,使用得到的粗粒子和溶劑,使用濕式介質分散型裝置進行粉碎,得到數均一次粒徑20nm的cuino2微粒。對于得到的cuino2微粒,通過與制造例1-1同樣的方法進行表面處理,制備了p型半導體微粒2。
[制造例1-3]p型半導體微粒3的制備
參考日本特開2014-170006號公報及日本特開2003-286096號公報,制作了srcu2o2微粒。具體地,將氧化鍶(sro)及氧化銅(cuo)以3:7的摩爾比混合,在氮中混入了5%以下的氧的氣氛下,在1000℃以上加熱熔融后,在保持在1000℃以上的狀態下,使由通式srcu2o2表示的種晶接觸,通過溶液提拉法(溶液ひきあげ法)得到srcu2o2單晶。將該單晶粉碎,進行篩分(分級),由此得到數均一次粒徑為20nm的srcu2o2微粒。對于得到的srcu2o2微粒,通過與制造例1-1同樣的方法進行表面處理,制備了p型半導體微粒3。
[制造例1-4]p型半導體微粒4的制備
除了將制造例1-3的氧化鍶(sro)變為氧化鋇(bao)以外,與制造例1-3同樣地得到數均一次粒徑為20nm的bacu2o2微粒。對于得到的bacu2o2微粒,通過與制造例1-1同樣的方法進行表面處理,制備了p型半導體微粒4。
[制造例1-5]p型半導體微粒5的制備
參考日本特開2015-129765號公報及日本特開2011-174167號公報,制作了cunbo微粒。具體地,將氧化銅(cu2o)及五氧化二鈮(nb2o5)以1:1的摩爾比充分地混合,接著,將該混合物的粉末使用錠劑成型機于35mpa壓縮成型,得到了氧化物的成型物。進而,在將該成型物放置在載于氧化鋁板上的上述的混合物的粉末上的狀態下,使用加熱到950℃的馬弗爐,在氮氣氛下燒結4小時而得到了燒結體。接著,使用得到的燒結體作為靶,使用脈沖激光蒸鍍裝置,在硼硅酸玻璃基板上使氧化物膜析出、生長,經過退火工序,由此得到了由nb和cu的復合氧化物形成的氧化物膜。然后,通過將該氧化物膜從硼硅酸玻璃基板分離、進行粉碎、分級,得到數均一次粒徑30nm的cu·nb復合氧化物(cunbo)微粒。對于得到的cunbo微粒,通過與制造例1-1同樣的方法進行表面處理,制備了p型半導體微粒5。
[制造例1-6]p型半導體微粒6的制備
除了將制造例1-5的五氧化二鈮(nb2o5)變為五氧化二鉭(ta2o5)以外,與制造例1-5同樣地得到數均一次粒徑30nm的cu·ta復合氧化物(cutao)微粒。對于得到的cutao微粒,通過與制造例1-1同樣的方法進行表面處理,制備了p型半導體微粒6。
[制造例1-7]p型半導體微粒7的制備
在制造例1-1中,除了將表面處理劑kbm-503變為3-丙烯酰氧基丙基三甲氧基硅烷“kbm-5103”(信越化學工業株式會社制造)以外,與制造例1-1同樣地制備了p型半導體微粒7。
[制造例1-8]n型半導體微粒1的制備
對于數均一次粒徑20nm的氧化錫(sno2)微粒(cikナノテック株式會社制造),通過與制造例1-1同樣的方法進行表面處理,制備了n型半導體微粒1。
<n型有機半導體的合成>
[制造例2-1]etm01的合成
在四口燒瓶中添加均苯四甲酸二酐300質量份、2-(三氟甲基)苯胺560質量份、及二甲基甲酰胺,在回流下加熱了3小時。冷卻后,將反應混合物過濾,用二甲基甲酰胺清洗沉淀物,進而用醚清洗、干燥。將該產物用硅膠柱精制,得到了下述化學式的化合物(etm01)。進行nmr、ir、ms、元素分析,確認得到目標物。
【化27】
[制造例2-2]etm02的合成
在制造例2-1中,除了將2-(三氟甲基)苯胺560質量份變為1,2-二甲基丙胺300質量份以外,與制造例2-1同樣地合成了下述化學式的化合物(etm02)。
【化28】
[制造例2-3]etm03的合成
在制造例2-1中,除了將均苯四甲酸二酐300質量份變為3,3’,4,4’-聯苯四羧酸二酐300質量份、且將2-(三氟甲基)苯胺560質量份變為2,6-二甲基苯胺310質量份以外,與制造例2-1同樣地合成了下述化學式的化合物etm03。
【化29】
[制造例2-4]etm04的合成
在制造例2-1中,除了將均苯四甲酸二酐300質量份變為萘-1,4,5,8-四羧酸二酐300質量份、且將2-(三氟甲基)苯胺560質量份變為4-氨基環己烷羧酸戊酯600質量份以外,與制造例2-1同樣地合成了下述化學式的化合物etm04。
【化30】
[制造例2-5]etm05的合成
在制造例2-1中,除了將均苯四甲酸二酐300質量份變為苝-3,4,9,10-四羧酸二酐300質量份、且將2-(三氟甲基)苯胺560質量份變為1-己基庚胺390質量份以外,與制造例2-1同樣地合成了下述化學式的化合物etm05。
【化31】
[制造例2-6]etm06的合成
(etm06a的合成)
在四口燒瓶中添加1,5-苯甲酰基萘60質量份及二甘醇,使其溶解,加入肼一水合物(80%)37.1質量份,加熱。在160℃下回流4小時后,冷卻到100℃,加入氫氧化鈉24質量份,加熱。進而在160℃~200℃下使其回流4小時,自然冷卻,使結晶析出。將析出的針狀結晶過濾分離,用甲醇清洗,進而用己烷/二氯甲烷=4/1(體積比)混合溶劑進行重結晶,得到了etm06a。
(etm06b的合成)
在四口燒瓶中添加馬來酸酐150質量份及硝基苯,在加熱回流下脫水。加入etm06a31質量份、碘0.5質量份,在200℃下加熱了5小時。然后,用減壓蒸餾將硝基苯進行回收至約2/3,自然冷卻。加入醋酸,使結晶析出,過濾分離。用醋酸和丙酮對過濾分離的粗結晶進行清洗,使其干燥,通過升華精制,得到了etm06b。
(etm06的合成)
在制造例2-1中,除了將均苯四甲酸二酐300質量份變為上述的etm06b300質量份、且將2-(三氟甲基)苯胺560質量份變為1,2-二甲基丙胺132質量份以外,與制造例2-1同樣地合成了下述化學式的化合物etm06。
【化32】
[制造例2-7]etm201的合成
etm201通過在以下所示的合成路線而合成。
【化33】
(etm201a的合成)
在四口燒瓶中加入[1,1’-聯苯]-4,4’-二酰氯10質量份、4-乙氧基苯甲酰肼13.9質量份、及無水吡啶,使其反應。將反應混合物投入純水中,將沉淀物過濾。將沉淀物用稀鹽酸、純水清洗后,用甲醇/乙醇混合溶劑進行重結晶,由此以針狀結晶的形式得到了etm201a。
(etm201的合成)
使etm201a15質量份在氧氯化磷812質量份中回流反應。將氧氯化磷餾除后,將反應產物用水、甲醇充分地清洗。接著將其用乙醇/甲苯混合溶劑進行重結晶,由此得到了etm201。
[制造例2-8]etm401的合成
etm401通過在以下所示的合成路線而合成。
【化34】
(etm401a的合成)
將根據參照文獻(j.org.chem.,2002,vol.67,9392-9396)合成的3-(2-溴苯基)吡啶23.4質量份溶解于醋酸,加入30%過氧化氫113.4質量份,在95℃下加熱攪拌5小時。將反應液在減壓下濃縮90%左右,在殘渣中加入1%小蘇打水,用醋酸乙酯將有機層抽出。用飽和食鹽水將有機層洗滌3次,將有機層在減壓下濃縮。在殘渣中加入氯仿,加入氧溴化磷43.0質量份,進行3小時加熱回流。將溶劑及過剩的氧溴化磷在減壓下餾除,將殘渣用甲苯進行重結晶,得到了etm401a。
(etm401b的合成)
在四口燒瓶中,使etm401a25.0質量份溶解于脫水乙醚中,將內溫冷卻到-75℃。接著,一邊將內溫保持在-70℃以下一邊緩慢地加入1.6m的正丁基鋰/己烷溶液75.1質量份。添加后,在相同溫度下攪拌2小時后,一邊保持內溫-65℃以下一邊緩慢地加入將二氯二苯基硅烷21.2質量份溶解于脫水乙醚的溶液。在相同溫度下攪拌3小時后,放置,加熱到室溫,進而進行2小時攪拌。反應結束后,將溶劑在減壓下餾除,將殘渣用二氯甲烷-乙醇混合溶劑進行重結晶,得到了etm401b。
(etm401c的合成)
在四口燒瓶中,使etm401b20.0質量份溶解于醋酸,加入30%過氧化氫67.6質量份,在95℃下加熱攪拌5小時。將反應液在減壓下濃縮90%左右,在殘渣中加入1%小蘇打水,用醋酸乙酯將有機層抽出。用飽和食鹽水將有機層清洗3次,將有機層在減壓下濃縮。在剩余物中加入氯仿,加入氧溴化磷25.6質量份,進行3小時加熱回流。將溶劑及過剩的氧溴化磷在減壓下餾除,將殘渣用甲苯進行重結晶,得到了etm401c。
(etm401的合成)
在四口燒瓶中,使etm401c23.0質量份溶解于dmac,加入咔唑34.1質量份、碘化銅23.3質量份、碳酸鉀16.9質量份、及l-脯氨酸1.3質量份,在氮氣流下、內溫150℃下進行6小時加熱攪拌。反應結束后,將溶劑在減壓下餾除,將殘渣用硅膠柱色譜(洗脫劑=庚烷:二氯甲烷=9:1(體積比))精制,得到了etm401的白色粉末。就結構而言,用核磁共振法(1h-nmr、13c-nmr等)及mass光譜(質量分析法)來鑒定。
[制造例2-9]etm501的合成
etm501通過在以下所示的合成路線而合成。
【化35】
(etm501a的合成)
在四口燒瓶中將茚三酮100質量份、及1,3-二苯基-2-丙酮118質量份添加到乙醇中,加熱直至進行回流。在回流的反應液中,歷時1小時滴加溶解了氫氧化鉀9.5質量份的甲醇溶液。滴加后,進行回流攪拌1小時,自然冷卻,使結晶析出,將結晶過濾分離,用甲醇清洗。用乙腈進行重結晶,得到了etm501a。
(etm501b的合成)
將etm501a130質量份、乙炔二羧酸二甲酯82.9質量份放入燒瓶中,將外溫設定為160℃,加熱反應液。內溫成為130℃以上后,加熱攪拌2小時后,將自然冷卻、析出的結晶過濾分離。在粗結晶中加入乙醇和甲苯,用重結晶得到了etm501b。
(etm501的合成)
在四口燒瓶中將etm501b155質量份、丙二腈45.7質量份加入到thf,在外溫-10℃下冷卻。在內溫0℃±5℃下滴加溶解于四氯化碳中的四氯化鈦328質量份。在這樣的溫度下攪拌了30分鐘后,添加吡啶274質量份。緩慢地回到室溫,進而加熱使其回流。在回流狀態下反應8小時后,回到室溫后,將反應液添加到純水中,進而加入甲苯,用分液將甲苯層抽出。將甲苯層用稀鹽酸清洗后,進行水洗直至水層的ph成為中性。用減壓蒸餾將甲苯層濃縮,加入甲苯-乙醇混合溶液,進行重結晶。將結晶過濾分離,再次進行重結晶,得到了etm501。
<電子照相感光體的制作>
[實施例1]
[導電性支承體]
對直徑80mm的鋁制的圓筒體的表面進行切削加工,準備了將表面細微地粗面化了的導電性支承體。通過在以下所示的方法,在該導電性支承體上依次層疊中間層、感光層及保護層,制作了電子照相感光體。
[中間層的制作]
(金屬氧化物微粒[1]的制備)
將數均一次粒徑為35nm的金紅石型氧化鈦“mt-500sa”(テイカ株式會社制造)500質量份、表面處理劑:3-甲基丙烯酰氧基丙基三甲氧基硅烷“kbm-503”(信越化學工業株式會社制造)65質量份、甲苯1500質量份攪拌混合后,通過珠磨機在磨機滯留時間25分鐘、溫度35℃下進行濕式粉碎處理,由得到的漿料通過減壓蒸餾將甲苯分離除去。通過將得到的干燥物在120℃下加熱2小時,進行了表面處理劑的燒結。然后,通過針磨機(ピンミル)進行粉碎、得到了被有機處理了的由金紅石型氧化鈦構成的金屬氧化物微粒[1]。
(金屬氧化物微粒[2]的制備)
在上述的金屬氧化物微粒[1]的制備中,除了作為表面處理劑代替3-甲基丙烯酰氧基丙基三甲氧基硅烷而使用了甲基氫聚硅氧烷(mhps):1,1,1,3,5,5,5-七甲基三硅氧烷(信越化學工業株式會社制造)以外,同樣地得到了被有機處理了的由金紅石型氧化鈦構成的金屬氧化物微粒[2]。
將由下述式表示的聚酰胺樹脂(n-1)100質量份加入乙醇/正丙醇/四氫呋喃(體積比45/20/35)的混合溶劑1700質量份中,在20℃下攪拌混合,在該溶液中添加上述的金屬氧化物微粒[1]97質量份及金屬氧化物微粒[2]226質量份,通過珠磨機,使磨機滯留時間為5小時來使其分散。將該溶液靜置一晝夜后,使用日本ポール株式會社制造的リジメッシュ5μm過濾器在50kpa的壓力下進行過濾,由此制備了中間層形成用涂布液。
用浸涂法將這樣得到的中間層形成用涂布液涂布于洗凈了的導電性支承體的外周面,在120℃下干燥30分鐘,由此形成了干燥膜厚2μm的中間層。
【化36】
[感光層的制作]
(電荷產生層的制作)
《顏料(cg-1)的合成》
(1)無定形氧鈦酞菁的合成
由1,3-二亞氨基異吲哚啉和四正丁氧基鈦合成粗氧鈦酞菁,將得到的粗氧鈦酞菁溶解于硫酸的溶液注入水中而使結晶析出。將該溶液過濾后,將得到的結晶用水充分地清洗,得到了濕糊。接著,在冷凍庫中使濕糊凍結,再次解凍后,進行過濾及干燥,由此得到了無定型氧鈦酞菁。
(2)(2r,3r)-2,3-丁二醇加成體氧鈦酞菁(cg-1)的合成
將得到的無定型氧鈦酞菁與(2r,3r)-2,3-丁二醇以(2r,3r)-2,3-丁二醇相對于無定型氧鈦酞菁的當量比成為0.6的方式在鄰二氯苯(odb)中混合。將得到的混合物在60~70℃下加熱攪拌6小時,將得到的溶液靜置一夜后,進一步添加甲醇而使結晶析出。將該溶液過濾后,將得到的結晶用甲醇清洗,由此得到了由含有(2r,3r)-2,3-丁二醇加成體氧鈦酞菁的顏料構成的電荷產生物質(cg-1)。
測定電荷產生物質(cg-1)的x射線衍射光譜,結果在8.3°、24.7°、25.1°、26.5°處確認有峰。推定得到的電荷產生物質(cg-1)為氧鈦酞菁與(2r,3r)-2,3-丁二醇的1:1加成體、和氧鈦酞菁(非加成體)的混合物。
《電荷產生層的形成》
將下述的成分混合、使用循環式超聲波均化器“rus-600tcvp”(株式會社日本精機制作所制造、19.5khz、600w),以循環流量40l/h分散0.5小時,制備了電荷產生層形成用涂布液:
·電荷產生物質(cg-1)24質量份
·聚乙烯醇縮丁醛樹脂エスレック(注冊商標)bl-1(積水化學工業株式會社制造)12質量份
·溶劑:甲乙酮/環己酮=4/1(v/v)400質量份。
在上述中間層上通過浸漬涂布法來涂布該電荷產生層形成用涂布液,形成涂布膜,將該涂布膜干燥,形成了層厚0.5μm的電荷產生層。
(電荷傳輸層的制作)
將電荷傳輸物質:4,4’-二甲基-4”-(β-苯基苯乙烯基)三苯基胺225質量份、粘結劑樹脂:聚碳酸酯樹脂“z300”(三菱ガス化學株式會社制造)300質量份、抗氧化劑:“irganox(注冊商標)1010”(basfジャパン株式會社制造)6質量份、溶劑:thf(四氫呋喃)1600質量份、溶劑:甲苯400質量份、硅油“kf-50”(信越化學工業株式會社制造)1質量份混合,溶解,制備了電荷傳輸層形成用涂布液。用浸涂法將該電荷傳輸層形成用涂布液在電荷產生層上涂布、形成了干燥膜厚20μm的電荷傳輸層。
[保護層的形成]
將下述的成分混合攪拌,充分地溶解·分散,制備了保護層形成用涂布液(固化性組合物):
使用圓形滑動料斗涂布裝置將該保護層形成用涂布液在上述的電荷傳輸層上涂布而形成涂布膜。將該涂布膜在室溫下干燥了20分鐘后,在氮流量為13.5l/分鐘的氮氣流下,使用氙燈作為光源,使該光源與涂布膜的表面的相距距離為5mm,在燈輸出功率4kw下照射1分鐘波長365nm的光(強度:4000mw/cm2、涂布膜中的光的照射強度:1800mw/cm2),由此形成層厚2.0μm的保護層,制作了感光體1。
[實施例2]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm02以外,同樣地制作了感光體2。
[實施例3]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm03以外,同樣地制作了感光體3。
[實施例4]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm04以外,同樣地制作了感光體4。
[實施例5]
在實施例1中,除了將保護層形成用涂布液的p型半導體微粒1變為p型半導體微粒3以外,同樣地制作了感光體5。
[實施例6]
在實施例5中,除了將保護層形成用涂布液的etm01變為etm05以外,同樣地制作了感光體6。
[實施例7]
在實施例2中,除了將保護層形成用涂布液的p型半導體微粒1變為p型半導體微粒2以外,同樣地制作了感光體7。
[實施例8]
在實施例2中,除了將保護層形成用涂布液的p型半導體微粒1變為p型半導體微粒4以外,同樣地制作了感光體8。
[實施例9]
在實施例2中,除了將保護層形成用涂布液的p型半導體微粒1變為p型半導體微粒5以外,同樣地制作了感光體9。
[實施例10]
在實施例1中,除了將保護層形成用涂布液的p型半導體微粒1變為p型半導體微粒6以外,同樣地制作了感光體10。
[實施例11]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm06以外,同樣地制作了感光體11。
[實施例12]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm201以外,同樣地制作了感光體12。
[實施例13]
實施例1中,除了將保護層形成用涂布液的etm01變為sigma-aldrich公司制造的3,3’,5,5’-四-叔丁基-4,4’-聯苯醌(etm311)以外,同樣地制作了感光體13。
【化37】
[實施例14]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm401以外,同樣地制作了感光體14。
[實施例15]
在實施例1中,除了將保護層形成用涂布液的etm01變為etm501以外,同樣地制作了感光體15。
[實施例16]
在實施例2中,除了將保護層形成用涂布液的p型半導體微粒1變為p型半導體微粒7以外,同樣地制作了感光體16。
[實施例17]
在實施例1中,除了在制備保護層形成用涂布液時將etm01的添加量變為5質量份、取而代之添加了5質量份etm02以外,同樣地制作了感光體17。
[實施例18]
在實施例2中,除了在制備保護層形成用涂布液時將p型半導體微粒1的添加量變為50質量份、取而代之添加了50質量份p型半導體微粒3以外,同樣地制作了感光體18。
[實施例19]
在實施例1中,除了制備保護層形成用涂布液時將p型半導體微粒1的添加量變為50質量份、取而代之添加了50質量份n型半導體微粒1以外,同樣地制作了感光體19。
[比較例1]
在實施例1中,除了制備保護層形成用涂布液時沒有添加etm01以外,同樣地制作了感光體20。
[比較例2]
在實施例1中,除了制備保護層形成用涂布液時沒有添加p型半導體微粒1以外,同樣地制作了感光體21。
[比較例3]
在實施例19中,除了在制備保護層形成用涂布液時沒有添加etm01以外,同樣地制作了感光體22。
<感光體的性能評價>
對于在實施例及比較例中得到的各感光體,進行了以下的評價。
[表面硬度]
使用超微小硬度計hm-2000(フィッシャー·インストルメンツ公司制造),測定了各感光體的表面硬度(通用硬度值)。具體地,將2mn的載荷施加于感光體表面10秒鐘,5秒的蠕變時間后,再次施加2mn的載荷10秒鐘,回復到初期狀態。如果膜硬度的值為150n/mm2以上,則作為感光體的耐久性沒有問題。
[圖像評價]
作為評價機,使用了將基本上具有圖1的構成的コニカミノルタビジネステクノロジーズ株式會社制“bizhubpro(注冊商標)c8000”的打印速度改造為120張/分鐘的評價機。將各感光體搭載于該評價機,評價了圖像性能(殘留電位·圖像記憶·點再現性·翳影)。
(殘留電位(δvi))
將感光體1~22分別搭載于上述評價機,首先,在溫度10℃、相對濕度20%rh的低溫低濕環境下,在黑色的位置將內部搭載圖案no.53/dot1(形成了具有規則性的點狀的曝光圖案的代表性的圖案)在濃度指示值255下100張連續地在轉印材料“podグロスコート”(a3尺寸、100g/m2)(王子制紙株式會社制造)上打印。算出了其第1張的曝光后電位與第100張的曝光后電位之差δvi1。接著,進行了50萬張連續地將相同條件的圖像進行打印的耐刷試驗后,進而,100張連續地打印了同條件的圖像。算出了其第1張的曝光后電位與第100張的曝光后電位之差δvi2。由δvi1和δvi2,按照下述的評價標準進行了評價。將結果示于表1中。
就曝光后電位而言,使用“cynthia59”(ジェンテック公司制造)、在溫度10℃、相對濕度15%rh的環境下進行測定。表面電位的變動的測定是一邊以130rpm使感光體旋轉一邊在柵電壓-800v、曝光量0.5μj/cm2的條件下反復地進行帶電和曝光而進行。
a:耐刷前后都為20v以下(合格)
b:耐刷前為20v以下,耐刷后為超過20v且45v以下(合格)
c:耐刷前為超過20v且40v以下、或者、耐刷前為20v以下且耐刷后為超過45v(不合格)
d:從耐刷前開始就超過40v(不合格)。
(圖像記憶)
將感光體1~22分別搭載于上述評價機,首先,在溫度10℃、相對濕度20%rh的低溫低濕環境下,在黑色的位置將內部搭載圖案no.53/dot1(形成了具有規則性的點狀的曝光圖案的代表性的圖案)在濃度指示值255下1000張連續地在轉印材料“podグロスコート”(a3尺寸、100g/m2)(王子制紙株式會社制造)上打印。
然后,10張連續地打印實心(ベタ)黑的部分和實心白的部分混在一起的白黑圖像,接著,印刷均勻的半色調圖像,對于該半色調圖像,以目視來觀察上述白黑圖像的經歷是否出現、即是否發生了記憶,按照下述的評價標準進行了評價(初期的評價)。
接著,進行了50萬張連續地打印同條件的圖像的耐刷試驗后,10張連續地打印實心黑的部分與實心白的部分混在一起的白黑圖像,接著印刷均勻的半色調圖像,對于該半色調圖像,以目視來觀察上述白黑圖像的經歷是否出現、即是否發生了記憶,按照下述的評價標準進行了評價(耐久后的評價)。
5:沒有發生圖像記憶(良好)
4:稀少地發生輕微的圖像記憶(實用上沒有問題)
3:能夠見到輕微的圖像記憶(實用上沒有問題)
2:稀少地發生不輕微的圖像記憶(實用上有問題)
1:發生不輕微的圖像記憶(實用上有問題)。
(點再現性)
在30℃/80%rh環境下,對于a3/podグロスコート紙(100g/m2、王子制紙株式會社制造),在濃度指示值100下1000張連續地兩面印刷了內部搭載圖案no.53/dot1(形成了具有規則性的點狀的曝光圖案的代表性的圖案)后,使用倍率100倍的放大鏡觀察點的形成狀態。接著,作為耐久試驗,50萬張連續地兩面印刷了同一圖案后,與上述同樣地對點的形成狀態進行了評價。予以說明,點再現性的判定標準如以下所述。
5:正常地形成了點(良好)
4:點稍微有些細小(實用上沒有問題)
3:點細小(實用上沒有問題)
2:幾乎沒有形成點(實用上有問題)
1:沒有形成點(實用上有問題)。
(翳影)
將感光體1~22分別搭載于上述評價機,首先,在溫度10℃、相對濕度20%rh的低溫低濕環境下,在黑色的位置將內部搭載圖案no.53/dot1(形成了具有規則性的點狀的曝光圖案的代表性的圖案)在濃度指示值255下1000張連續地在轉印材料“podグロスコート”(a3尺寸、100g/m2)(王子制紙株式會社制造)上打印。
然后,將沒有形成圖像的轉印材料“podグロスコート”(a3尺寸、100g/m2)(王子制紙株式會社制造)輸送到黑色的位置,在柵電壓-800v、顯影偏壓-650v的條件下形成不帶花紋圖像(白色實心圖像),以目視觀察得到的轉印材料上的翳影的有無。同樣地,在柵電壓-800v、顯影偏壓-650v的條件下形成黃色實心圖像,以目視觀察得到的轉印材料上的翳影的有無。然后,按照以下的評價標準進行了評價(初期的評價)。
接著,進行了50萬張連續地打印同條件的圖像的耐刷試驗后,將沒有形成圖像的轉印材料“podグロスコート”(a3尺寸、100g/m2)(王子制紙株式會社制造)輸送到黑色的位置,在柵電壓-800v、顯影偏壓-650v的條件下形成不帶花紋圖像(白色實心圖像),以目視觀察得到的轉印材料上的翳影的有無。同樣地,在柵電壓-800v、顯影偏壓-650v的條件下形成黃色實心圖像,以目視觀察了得到的轉印材料上的翳影的有無。然后,按照以下的評價標準進行了評價(耐久后的評價)。
5:對于白色實心圖像、黃色實心圖像這兩者,沒有觀察到翳影(合格)
4:對于白色實心圖像、黃色實心圖像的任一者,如果放大,則少量地觀察到翳影(實用上沒有問題)
3:對于白色實心圖像、黃色實心圖像這兩者,如果放大,則觀察到翳影(實用上沒有問題)
2:對于白色實心圖像、黃色實心圖像的任一者,以目視少量地觀察到翳影(實用上有問題)
1:對于白色實心圖像、黃色實心圖像的任一者,顯著地觀察到翳影(實用上有問題)。
將實施例及比較例的感光體的構成及評價結果示于下述表1中。
【表1】
- 還沒有人留言評論。精彩留言會獲得點贊!