光輝性色調劑、靜電荷圖像顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法與流程
本發明涉及光輝性色調劑、靜電荷圖像顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法。
背景技術:
近年來,檢驗了出于形成具有光澤(例如金屬光澤)的圖像的目的而使用含有光輝性顏料的光輝性色調劑。
因此,為了提供在電子照相印刷工序中重現金或銀色調的色調劑,公開了一種色調劑,其具有金屬色調并含有金屬顏料,其中所述金屬顏料含有:a)選自脂肪酸、至少一種酸的酰胺、至少一種酸的鹽、烯烴材料、天然蠟、合成蠟、聚合物及其組合的有機層(所述有機層含有電荷控制劑以及必要時的著色劑);以及必要時的b)硅酸鹽、鈦酸鹽或鋁酸鹽的涂布膜,并且必要時用疏水性氣相法金屬氧化物(fumedmetaloxide)覆蓋色調劑(例如,參見專利文獻1)。
為了提供防止含板狀金屬顏料的色調劑的清潔故障的圖像形成設備,公開了一種圖像形成設備,其包含:圖像保持部件;充電單元,其對圖像保持部件的表面充電;靜電荷圖像形成單元,其在圖像保持部件的經充電表面上形成靜電荷圖像;顯影單元,其容納靜電荷圖像顯影劑并通過利用靜電荷圖像顯影劑將形成在圖像保持部件表面上的靜電荷圖像顯影為色調劑圖像,所述靜電荷圖像顯影劑含有平均縱向長度為5μm~12μm且平均厚度為0.01μm~0.5μm的板狀金屬顏料,并且含有平均縱向長度為7μm~20μm、平均厚度為1μm~3μm且平均圓形度為0.5~0.9的扁平狀色調劑;轉印單元,其將形成在圖像保持部件表面上的色調劑圖像轉印至記錄介質的表面上;校準單元,其使轉印后殘留在圖像保持部件表面上的色調劑從圖像保持部件表面上升起;清潔單元,其包含用于清潔轉印后殘留在圖像保持部件表面上的色調劑的清潔刮板;和定影單元,其將轉印至記錄介質表面上的色調劑圖像定影(例如,參見專利文獻2)。
[專利文獻1]jp-t-2009-501349
[專利文獻2]jp-a-2015-118358
技術實現要素:
本發明的示例性實施方式的一個目的是提供一種光輝性色調劑,其包含含有粘合劑樹脂和光輝性顏料的色調劑顆粒,并且相比于光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形,所述光輝性色調劑進一步防止在形成灰度圖像(gradationimages)的情形中的顆粒度劣化。
上述目的通過以下構造實現。
根據本發明的第一方面,提供了一種光輝性色調劑,其包含:
含有粘合劑樹脂和光輝性顏料的色調劑顆粒,
其中所述光輝性顏料的由jisk5906:2009定義的水面擴散面積為5m2/g~20m2/g,并且
所述光輝性顏料的平均縱向長度為0.5μm~10μm。
根據本發明的第二方面,在第一方面所述的光輝性色調劑中,所述光輝性顏料的平均縱向長度等于或大于0.5μm且小于5μm。
根據本發明的第三方面,在第一方面所述的光輝性色調劑中,所述光輝性顏料是金屬顏料。
根據本發明的第四方面,在第三方面所述的光輝性色調劑中,所述金屬顏料是鋁顏料。
根據本發明的第五方面,在第一方面所述的光輝性色調劑中,所述色調劑顆粒的體積平均粒徑為3μm~30μm。
根據本發明的第六方面,在第一方面所述的光輝性色調劑中,所述色調劑顆粒的長徑比為1.5~15。
根據本發明的第七方面,在第一方面所述的光輝性色調劑中,所述光輝性色調劑的平均縱向長度(lp)與所述光輝性色調劑的體積平均粒徑(dt)之比(lp/dt)為0.017~1.000。
根據本發明的第八方面,在第一方面所述的光輝性色調劑中,相對于全部色調劑,所述色調劑中除無機物之外的甲苯不溶性部分為0.1重量%~50重量%。
根據本發明的第九方面,在第一方面所述的光輝性色調劑中,所述色調劑的平均最大厚度c與平均圓當量直徑d之比(c/d)為0.001~0.700。
根據本發明的第十方面,在第一方面所述的光輝性色調劑中,所述粘合劑樹脂含有脲改性聚酯樹脂。
根據本發明的第十一方面,提供了一種靜電荷圖像顯影劑,其包含:
第一~第十方面中任一項所述的光輝性色調劑。
根據本發明的第十二方面,提供了一種色調劑盒,其包含:
含有第一~第十方面中任一項所述的光輝性色調劑的容器,所述色調劑盒能夠從圖像形成設備上拆卸。
根據本發明的第十三方面,提供了一種處理盒,其包含:
顯影單元,所述顯影單元容納第十一方面所述的靜電荷圖像顯影劑,并通過利用所述靜電荷圖像顯影劑使圖像保持部件的表面上形成的靜電荷圖像顯影為色調劑圖像,
其中所述處理盒能夠安裝到圖像形成設備上并從圖像形成設備上拆卸。
根據本發明的第十四方面,提供了一種圖像形成設備,其包含:
圖像保持部件;
充電單元,其對所述圖像保持部件的表面充電;
靜電荷圖像形成單元,其在所述圖像保持部件的經充電表面上形成靜電荷圖像;
顯影單元,其容納第十一方面所述的靜電荷圖像顯影劑并通過利用所述靜電荷圖像顯影劑使所述圖像保持部件的表面上形成的所述靜電荷圖像顯影為色調劑圖像;
轉印單元,其將所述圖像保持部件的表面上形成的所述色調劑圖像轉印至記錄介質的表面;和
定影單元,其將轉印到所述記錄介質的表面的所述色調劑圖像定影。
根據本發明的第十五方面,提供了一種圖像形成方法,其包括:
對圖像保持部件的表面充電;
在所述圖像保持部件的經充電表面上形成靜電荷圖像;
通過利用第十一方面所述的靜電荷圖像顯影劑將所述圖像保持部件的表面上形成的所述靜電荷圖像顯影為色調劑圖像;
將所述圖像保持部件的表面上形成的所述色調劑圖像轉印至記錄介質的表面上;和
將轉印到所述記錄介質的表面上的所述色調劑圖像定影。
根據本發明的第一方面和第七~第九方面,提供了一種光輝性色調劑,與光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形相比,所述光輝性色調劑進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第二方面,與光輝性顏料的平均縱向長度小于0.5μm或等于或大于5μm的情形相比,進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第三方面,與使用除金屬顏料之外的顏料作為光輝性顏料的情形相比,進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第四方面,與使用除鋁顏料之外的顏料作為金屬顏料的情形相比,進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第五方面,與色調劑顆粒的體積平均粒徑小于3μm或大于30μm的情形相比,進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第六方面,與色調劑顆粒的長徑比小于1.5或大于15的情形相比,進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第十方面,與粘合劑樹脂不含有脲改性聚酯樹脂的情形相比,易于防止由光輝性顏料造成的在定影部件上損傷的發生。
根據本發明的第十一方面,提供了一種包含光輝性色調劑的靜電荷圖像顯影劑,與光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形相比,所述光輝性色調劑進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第十二方面,提供了一種容納光輝性色調劑的色調劑盒,與光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形相比,所述光輝性色調劑進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第十三方面,提供了一種容納含有光輝性色調劑的靜電荷圖像顯影劑的處理盒,與光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形相比,所述光輝性色調劑進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第十四方面,提供了一種使用含有光輝性色調劑的靜電荷圖像顯影劑的圖像形成設備,與光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形相比,所述光輝性色調劑進一步防止在形成灰度圖像的情形中的顆粒度劣化。
根據本發明的第十五方面,提供了一種使用含有光輝性色調劑的靜電荷圖像顯影劑的圖像形成方法,與光輝性顏料的由jisk5906:2009定義的水面擴散面積小于5m2/g或大于20m2/g或者平均縱向長度小于0.5μm或大于10μm的情形相比,所述光輝性色調劑進一步防止在形成灰度圖像的情形中的顆粒度劣化。
附圖說明
將基于以下附圖詳細描述本發明的示例性實施方式,其中:
圖1是示意性圖解示例性實施方式的色調劑顆粒的實例的截面圖;
圖2是示意性圖解示例性實施方式的圖像形成設備的實例的構造圖;和
圖3是示意性圖解示例性實施方式的處理盒的實例的構造圖。
具體實施方式
下文中,將提供本發明示例性實施方式的光輝性色調劑、靜電荷圖像顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法的詳述。
光輝性色調劑
本示例性實施方式的光輝性色調劑(下文中在一些情形中也稱作“色調劑”)是包含色調劑顆粒的色調劑,所述色調劑顆粒包含粘合劑樹脂和光輝性顏料,其中所述光輝性顏料的由jisk5906:2009定義的水面擴散面積(下文中在一些情形中也稱作“水面擴散面積”)為5m2/g~20m2/g,且平均縱向長度為0.5μm~10μm。
根據示例性實施方式的色調劑,防止在形成灰度圖像的情形中的顆粒度劣化。此原因雖不清楚,但推測如下。
在用光輝性色調劑形成灰度圖像的情形中,特別是當在高溫和高濕度環境下高速形成大量灰度圖像時,灰度圖像中的點干擾可在灰度圖像中分別導致存在光輝性顏料的部分和不存在光輝性顏料的部分。灰度圖像中的點干擾在視覺上認定為灰度圖像中的顆粒度劣化。由于相關領域中的光輝性色調劑具有較大粒徑,且每單位重量色調劑的覆蓋記錄介質(如紙)的面積小,故易于在灰度圖像中出現未被光輝性顏料覆蓋的間隙。這被認為是灰度圖像中易于出現點干擾的原因。由于光輝性色調劑一般具有較大粒徑,故不存在光輝性顏料的部分醒目地出現。
根據本示例性實施方式的色調劑,使用由jisk5906:2009定義的水面擴散面積為5m2/g~20m2/g的光輝性顏料作為所述光輝性顏料。因此,每單位重量色調劑的覆蓋記錄介質的面積較大。因此,即使在將光輝性色調劑轉印至記錄介質表面上時的點在灰度圖像中被干擾時,由于在光輝性色調劑的定影期間在灰度圖像中不易出現未被光輝性顏料覆蓋的間隙,故易于獲得光滑的灰度圖像。因此,認為防止在形成灰度圖像的情形中的顆粒度劣化。
在示例性實施方式中,“顆粒度”是表明圖像缺陷的尺度,并且隨著顆粒度提高,圖像缺陷降低。
在本示例性實施方式的色調劑中,相對于全部色調劑,無機物之外的甲苯不溶性部分優選為0.1重量%~50重量%,更優選為2重量%~30重量%,進一步優選為3重量%~10重量%。
例如,通過以下方法調節甲苯不溶性部分:1)通過向在末端具有反應性官能團的聚合物成分添加交聯劑而形成交聯結構或支化結構的方法;2)通過向在末端具有離子型官能團的聚合物成分添加多價金屬離子而形成交聯結構或支化結構的方法;或3)通過用異氰酸酯等進行處理而延長或形成樹脂鏈長度的分支的方法。
在此,甲苯不溶性部分是色調劑組分中除無機物之外的組分,其不溶于甲苯。然而,在色調劑顆粒中含有防粘劑以及光輝性顏料和粘合劑樹脂的情形中,所述甲苯不溶性部分表示無機物和防粘劑之外的甲苯不溶部分。換言之,所述甲苯不溶部分表示含有不溶于甲苯的粘合劑樹脂成分(具體而言,粘合劑樹脂的高分子量成分)作為主要成分(例如,相對整體而言等于或大于90重量%)的不溶性部分。非光輝性顏料和外添劑等以及光輝性顏料對應于無機物。
甲苯不溶性部分是通過以下方法測量的值。
首先,用雙酚a型液體環氧樹脂和固化劑包埋作為測量目標的色調劑,然后形成切割用樣品。隨后,使用利用金剛石刀具的切割機(例如ultracutuct(由leica制造))在100℃將樣品切割成片。
通過具有能量分散型x射線分析儀(sem-edx)的掃描電子顯微鏡觀察切割成片的樣品截面,并且通過能量分散型x射線分析儀(edx)識別存在于色調劑中的無機物(扁平狀光輝性顏料(在截面中以針狀觀察到)和在對色調劑顆粒外部添加外添劑的情形中的外添劑)的組成元素。隨后,通過熒光x射線分析儀確定無機物的量(重量%)。
在此,使用具有能量分散型x射線分析儀“emax模型6923h”(由horiba,ltd.制造)的電子顯微鏡“s-4100”(由horiba,ltd.制造)作為所述具有能量分散型x射線分析儀的掃描電子顯微鏡,并且作為測量條件將加速電壓設定為20kv。相比之下,使用由shimadzucorporation制造的“熒光x射線分析儀xrf-1500”作為所述熒光x射線分析儀,并且作為測量條件將管電壓設定為40kv,將管電流設定為90ma,并將測量時間設定為5分鐘。
相比之下,將1g稱量的色調劑放入由稱量的玻璃纖維制成的圓柱形濾紙中,并附接至加熱型索氏提取器的提取管。隨后,將甲苯倒入燒瓶中并利用覆套式加熱器以110℃加熱。另外,利用附接至提取管的加熱器以125℃加熱提取管的外圍。以一定的回流速度進行提取,以便一次提取循環在4分鐘~5分鐘內進行。在提取10小時之后,提取圓柱形濾紙和色調劑殘留物,干燥并稱重。
然后,基于下式計算色調劑殘留量(重量%):色調劑殘留量(重量%)=[(圓柱形濾紙量+色調劑殘留量)(g)–柱形濾紙量(g)]/色調劑重量(g)×100。色調劑殘留物包含無機物(如光輝性顏料和外添劑)以及甲苯不溶性部分。在色調劑顆粒含有防粘劑的情形中,由于通過加熱進行了提取,防粘劑對應于甲苯可溶性部分。
隨后,由通過熒光x射線分析儀量化的“無機物(光輝性顏料和在外部添加外添劑的情形中的外添劑)的量(重量%)”和通過加熱索氏提取器而提取的“色調劑殘留量(重量%)”計算甲苯不溶性部分(重量%)。換言之,由等式“甲苯不溶性部分(重量%)”=“色調劑殘留量(重量%)”–“無機物的量(重量%)”計算甲苯不溶性部分(重量%)。
在本示例性實施方式的色調劑中,“光輝性”是指在通過光輝性色調劑形成圖像時所觀察到的光澤,如金屬光澤。
具體而言,本示例性實施方式的色調劑優選具有2~100的比例(x/y),其中,在形成實心圖像并用入射光以–45°的入射角照射該圖像的情形中,通過變角光度計測量受光角+30°處的反射率x和受光角-30°處的反射率y。
等于或大于2的比例(x/y)表示與入射側相對側(正角側)上的反射大于入射光入射的入射側(負角側)上的反射,即,防止了入射光的漫反射。在入射光的漫反射在各方向上發生的情形中,在視覺識別中觀察到反射光具有暗色調的顏色。因此,如果比例(x/y)小于2,則存在在反射光的視覺識別中不能觀察到光澤并且光輝性劣化的情形。
相比之下,如果比例(x/y)超過100,則存在以下情形,可視覺識別反射光的視角變得極其狹窄,并且由于正反射光成分大,根據視角會觀察到黯黑顏色。
就光輝性和色調劑制造性的角度而言,比例(x/y)優選為4~50,更優選為6~20,特別優選為8~15。
通過變角光度計對比例(x/y)的測定
在此,首先將給出入射角和受光角的描述。在本示例性實施方式中,在通過變角光度計進行測量時,將入射角設定為-45°,這是因為對于具有較寬范圍的光澤度的圖像而言實現了高測量靈敏度。
將受光角設定為-30°和+30,這是由于在有光澤感圖像和無光澤感圖像的評估中實現了最高的測量靈敏度。
接下來,將給出測量比例(x/y)的方法的描述。
使用由nippondenshokuindustriesco.,ltd.制造的分光式變角色差儀gc5000l作為可變角光度計,從而以相對于作為測量目標的圖像(光輝性圖像)為-45°的入射角使入射光入射在所述圖像上,并且測量受光角+30°處的反射率x和受光角-30°處的反射率y。以20nm的間隔由400nm~700nm的波長范圍內的光測量反射率x和反射率y,并且獲得各波長處的反射率的平均值。由這些測量結果計算比例(x/y)。
從滿足上述比例(a/b)的角度而言,本示例性實施方式的色調劑優選滿足以下要求(1)和(2)。
(1)平均圓當量直徑d大于色調劑顆粒的平均最大厚度c。
(2)在色調劑顆粒厚度方向上的截面的縱軸方向與光輝性顏料在其觀察的縱軸方向之間角度為-30°~+30°的光輝性顏料的比例等于或小于觀察到的全部光輝性顏料的60%。
據認為如果色調劑顆粒具有圓當量直徑長于厚度的扁平形狀(參見圖1),則在形成圖像的定影工序中,在定影期間由于壓力,扁平狀色調劑顆粒的扁平表面側排列為面向記錄介質表面。在圖1中,2表示色調劑顆粒,4表示光輝性顏料,而l表示色調劑顆粒的厚度。
因此,據認為在色調劑中所含的扁平狀(薄片狀)光輝性顏料顆粒中滿足上述要求(2)“色調劑截面的縱軸方向與光輝性顏料的縱軸方向之間的角度為-30°~+30°”的光輝性顏料顆粒的最大面積表面側排列為面向記錄介質的表面。據認為由于在使光入射在由此形成的圖像上的情形中,針對入射光,以漫射形式反射的光輝性顏料的比例被防止,故實現了上述范圍的比例(x/y)。
下文中,將給出本示例性實施方式的色調劑的詳細描述。
本示例性實施方式的色調劑包含色調劑顆粒,其至少含有粘合劑樹脂和光輝性顏料。根據需要,本示例性實施方式的色調劑顆粒可含有其他成分。
本示例性實施方式的色調劑可包含含有光輝性顏料和粘合劑樹脂的色調劑顆粒,以及對該色調劑顆粒外部添加的外添劑。
色調劑顆粒
所述色調劑顆粒含有粘合劑樹脂和光輝性顏料。根據需要,所述所述色調劑顆粒可含有其他添加劑,例如防粘劑。
粘合劑樹脂
粘合劑樹脂的實例包括諸如以下單體的均聚物或這些單體中的兩種以上類型的共聚物的乙烯基樹脂,該單體例如:苯乙烯類(如苯乙烯、對氯苯乙烯或α-甲基苯乙烯);(甲基)丙烯酸酯類(如丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丙酯、丙烯酸正丁酯、丙烯酸十二烷基酯、丙烯酸2-乙基己酯、甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸正丙基、甲基丙烯酸十二烷基酯或甲基丙烯酸2-乙基己酯);烯鍵式不飽和腈類(如丙烯腈或甲基丙烯腈);乙烯基醚類(如乙烯基甲基醚或乙烯基異丁基醚);乙烯基酮類(如乙烯基甲基酮、乙烯基乙基酮或乙烯基異丙烯基酮);或烯烴類(如乙烯、丙烯或丁二烯)。
粘合劑樹脂的實例還包括非乙烯基樹脂,例如環氧樹脂、聚酯樹脂、聚氨酯樹脂、聚酰胺樹脂、纖維素樹脂、聚醚樹脂或改性松香,此類非乙烯基樹脂與所述乙烯基樹脂的混合物,以及通過所述非乙烯基樹脂的共存在使乙烯基單體聚合而獲得的接枝聚合物。
此類粘合劑樹脂可以單獨使用一種,或組合使用兩種以上。
優選使用聚酯樹脂作為粘合劑樹脂。
聚酯樹脂的實例包括已知聚酯樹脂。
聚酯樹脂的實例包括多元羧酸和多元醇的縮聚物。可以使用商購聚酯樹脂或合成聚酯樹脂。
多元羧酸的實例包括脂肪族二羧酸(例如,草酸、丙二酸、馬來酸、富馬酸、檸康酸、衣康酸、戊烯二酸、琥珀酸、烯基琥珀酸酯、己二酸或癸二酸)、脂環族二羧酸(例如,環己烷二羧酸)、芳香族二羧酸(例如,對苯二甲酸、間苯二甲酸、鄰苯二甲酸或萘二甲酸)、它們的酸酐或其低級烷基酯(例如含有1~5個碳原子)。在所述實例中,例如,優選使用芳香族二羧酸作為多元羧酸。
作為多元羧酸,可以與二元羧酸組合使用具有交聯結構或支化結構的三元以上的羧酸。三元以上的羧酸的實例包括偏苯三酸、均苯四酸、它們的酸酐或其低級烷基酯(例如含有1~5個碳原子)。
多元羧酸的一種或兩種以上可單獨或組合使用。
多元醇的實例包括脂肪族二醇(例如,乙二醇、二乙二醇、三乙二醇、丙二醇、丁二醇、己二醇或新戊二醇)、脂環族二醇(例如,環己二醇、環己烷二甲醇或氫化雙酚a)、芳香族二醇(如雙酚a的氧化乙烯加合物或雙酚a的氧化丙烯加合物)。在所述實例中,作為所述多元醇,優選使用芳香族二醇、脂環族二醇,更優選使用芳香族二醇。
作為多元醇,可以與二醇組合使用具有交聯結構或支化結構的三元以上的多元醇。三元以上的多元醇的實例包括甘油、三羥甲基丙烷和季戊四醇。
多元醇的一種或兩種以上可單獨或組合使用。
聚酯樹脂的玻璃化轉變溫度(tg)優選為50℃~80℃,更優選為50℃~65℃。
由通過差示掃描量熱法(dsc)獲得的dsc曲線來確定玻璃化轉變溫度。更具體而言,基于jisk7121-1987“塑料轉變溫度的測試方法”中如何獲得玻璃化轉變溫度的方法中描述的“外推法玻璃化轉變起始溫度”來測定玻璃化轉變溫度。
聚酯樹脂的重均分子量(mw)優選為5,000~1,000,000,更優選為7,000~500,000。
聚酯樹脂的數均分子量(mn)優選為2,000~100,000。
聚酯樹脂的分子量分布mw/mn優選為1.5~100,更優選為2~60。
通過凝膠滲透色譜法(gpc)測量重均分子量和數均分子量。使用作為測量設備的由tosohcorporation制造的gpc·hlc-8120gpc、由tosohcorporation制造的柱tskgelsuperhm-m(15cm)和thf溶劑進行gpc分子量測量。使用由單分散苯乙烯標準樣品形成的分子量校準曲線從所述測量結果計算重均分子量和數均分子量。
通過已知制造方法獲得聚酯樹脂。具體而言,通過將聚合溫度設定為180℃~230℃獲得聚酯樹脂,例如,根據需要降低反應體系中的壓力,并且使在除去縮合期間生成的水和醇的同時進行引起反應。
在原料的單體在反應溫度下不溶解或不共混的情形中,可添加具有高沸點的溶劑作為增溶劑來促進溶解。在此情形中,縮聚反應在蒸發增溶劑的同時進行。在共聚反應中存在低相容性單體的情形中,優選的是預先使低相容性的單體和酸或醇縮合以便與該單體縮聚,并且隨后與主要組分進行縮聚。
在此,上述未改性聚酯樹脂之外的聚酯樹脂的實例包括改性聚酯樹脂。改性聚酯樹脂是其中存在酯連接之外的連接基團的聚酯樹脂,或者是其中與聚酯樹脂成分不同的樹脂成分通過共價鍵或離子鍵鍵合的聚酯樹脂。改性聚酯的實例包括通過以下方式制備的具有如下進行改性的末端的樹脂:使在末端導入有與酸基或羥基反應的官能團(例如異氰酸酯基團)的聚酯樹脂與活性氫化合物反應。
特別優選脲改性聚酯樹脂作為改性樹脂。包含脲改性聚酯樹脂作為粘合劑樹脂利于防止定影部件中出現由于光輝性顏料造成的損壞。這被認為是由于脲改性聚酯具有適當的親水性并設置在色調劑顆粒中,從而作為甲苯不溶性部分圍繞光輝性顏料的邊緣部分。就這點而言,相對于全部粘合劑樹脂,脲改性聚酯樹脂的含量優選為10重量%~30重量%,更優選為15重量%~25重量%。
作為脲改性聚酯樹脂,使用通過具有異氰酸酯基團的聚酯樹脂(聚酯預聚物)和胺化合物之間的反應(交聯反應和伸長反應中的至少一個)獲得的脲改性聚酯樹脂。脲改性聚酯可含有氨基甲酸酯鍵和脲鍵。
具有異氰酸酯基團的聚酯預聚物是多元羧酸與多元醇的縮聚物,其實例包括通過引起具有活性氫的聚酯和多價異氰酸酯化合物之間的反應而獲得的預聚物。聚酯中包含的具有活性氫的基團的實例包括羥基(醇式羥基和酚式羥基)、氨基、羧基和巰基,優選使用醇式羥基。
具有異氰酸酯基團的聚酯預聚物中的多元羧酸和多元醇的實例包括與針對聚酯樹脂描述的那些多元羧酸和多元醇相似的化合物。
多價異氰酸酯化合物的實例包括:脂肪族聚異氰酸酯(例如四亞甲基二異氰酸酯、六亞甲基二異氰酸酯或2,6-二異氰酸酯甲基己酸酯);脂環族聚異氰酸酯(例如異佛爾酮二異氰酸酯、環己基甲烷二異氰酸酯);芳香族二異氰酸酯(甲代亞苯基(tolylene)二異氰酸酯或二苯甲烷二異氰酸酯);芳香族-脂肪族二異氰酸酯(α,α,α’,α’-四甲基亞二甲苯基二異氰酸酯);異氰尿酸酯;以及通過諸如酚衍生物、肟或己內酰胺等封阻劑將上述聚異氰酸酯封阻而制備的那些。
對于多價異氰酸酯化合物,可單獨使用一種或組合使用兩種以上的封阻劑。
針對多價異氰酸酯化合物的比例,具有羥基的聚酯預聚物中的異氰酸酯基團[nco]和羥基[oh]之間的當量比[nco]/[oh]優選為1/1~5/1,更優選為1.2/1~4/1,進一步優選為1.5/1~2.5.1。如果[nco]/[oh]設定在1/1~5/1,則甲苯不溶性部分在上述范圍內的趨勢增加,并且定影部件中出現的由于光輝性顏料造成的損壞易于被抑制。如果[nco]/[oh]設定為等于或小于5,則易于防止低溫定影性的劣化。
在具有異氰酸酯基團的聚酯預聚物中,相對于全部具有異氰酸酯基團的聚酯預聚物,源自于多價異氰酸酯化合物的成分的含量優選為0.5重量%~40重量%,更優選為1重量%~30重量%,進一步優選為2重量%~20重量%。如果源自于多價異氰酸酯化合物的成分的含量被設定為0.5重量%~40重量%,則甲苯不溶性部分在上述范圍內的趨勢增加,并且定影部件中出現的由于光輝性顏料造成的損壞易于被抑制。如果源自于多價異氰酸酯化合物的成分的含量被設定為等于或小于40重量%,則易于防止低溫定影性的劣化。
具有異氰酸酯基團的聚酯預聚物的一個分子中所含的異氰酸酯基團數優選平均等于或大于1,更優選為平均1.5~3,進一步優選為平均1.8~2.5。如果每個分子的異氰酸酯基團數被設定為等于或大于1,則反應后的脲改性聚酯樹脂的分子量增加,甲苯不溶性部分在上述范圍內的趨勢增加,并且易于防止定影部件中出現的由于光輝性顏料造成的損壞。
待與具有異氰酸酯基團的聚酯預聚物反應的胺化合物的實例包括二胺、三元以上的多胺、氨基醇、氨基硫醇、氨基酸以及通過將其氨基封阻而獲得的化合物。
二胺的實例包括芳香族二胺(亞苯基二胺、二乙基甲苯二胺或4,4’-二氨基苯甲烷);脂環族二胺(例如4,4-二氨基-3,3’-二甲基二環己基甲烷、二胺環己烷或異佛爾酮二胺);和脂肪族二胺(例如乙二胺、四亞甲基二胺或六亞甲基二胺)。
三元以上的多胺的實例包括二亞乙基三胺和三亞乙基四胺。
氨基醇的實例包括乙醇胺和羥乙基苯胺。
氨基硫醇的實例包括氨乙基硫醇和氨丙基硫醇。
氨基酸的實例包括丙氨酸和氨基己酸。
通過將其氨基封阻而獲得的化合物的實例包括由胺化合物(例如二胺、三元以上的多胺、氨基醇、氨基硫醇或氨基酸)和酮化合物(例如丙酮、甲基乙基酮或甲基異丁基酮)獲得的酮亞胺化合物以及噁唑啉化合物。
在這些胺化合物中,優選使用酮亞胺化合物。
可單獨使用一種或組合使用兩種以上的胺化合物。
脲改性聚酯樹脂可以是通過如下方式在反應后具有調節過的分子量的樹脂:為了停止交聯反應和伸長反應中的至少一個,用終止劑(下文中也稱作“交聯/伸長反應終止劑”)調節具有異氰酸酯基團的聚酯樹脂(聚酯預聚物)和胺化合物之間的反應(交聯反應和伸長反應中的至少一個)。
交聯/伸長反應終止劑的實例包括一元胺(例如二乙胺、二丁胺、丁胺或月桂胺)和通過將一元胺封阻而獲得的物質(酮亞胺化合物)。
對于胺化合物的比例,具有異氰酸酯基團的聚酯預聚物中的異氰酸酯基團[nco]和胺中氨基[nhx]的當量比[nco]/[nhx]優選為1/2~2/1,更優選為1/1.5~1.5/1,進一步優選為1/1.2~1.2/1。如果[nco]/[nhx]設定在上述范圍內,則反應后的脲改性聚酯樹脂的分子量增加,甲苯不溶性部分在上述范圍內的趨勢增加,并且易于防止定影部件中出現的由于光輝性顏料造成的損壞。
脲改性聚酯樹脂的玻璃化轉變溫度優選為40℃~65℃,進一步優選為45℃~60℃。數均分子量優選為2,500~50,000,進一步優選為2,500~30,000。重均分子量為10,000~500,000,進一步優選為30,000~100,000。
例如,相對于全部色調劑顆粒,粘合劑樹脂的含量優選為40重量%~95重量%,更優選為50重量%~90重量%,進一步優選為60重量%~85重量%。
光輝性顏料
作為本示例性實施方式中所用的光輝性顏料,可使用由jisk5906:2009定義的水面擴散面積為5m2/g~20m2/g且平均縱向長度為0.5μm~10μm的任何顏料而無特別限制。
如果光輝性顏料的水面擴散面積小于5m2/g,則每單位重量色調劑的覆蓋記錄介質(例如紙)的面積小。因此,在形成灰度圖像的情形中的顆粒度可能劣化。相比之下,如果水面擴散面積大于20m2/g,則顏料的取向可能被干擾,且在形成灰度圖像的情形中的顆粒度可能劣化。本示例性實施方式中所用的光輝性顏料優選具有6m2/g~15m2/g的水面擴散面積,更優選具有7m2/g~10m2/g的水面擴散面積。
如果光輝性顏料的平均縱向長度小于0.5μm,則顏料的取向可能被干擾,且在形成灰度圖像的情形中的顆粒度可能劣化。相比之下,如果平均縱向長度大于10μm,則各光輝性顏料的亮度可能增加,并且顆粒度可能劣化。本示例性實施方式中所用的光輝性顏料的平均縱向長度優選等于或大于0.5μm且小于5μm,更優選為1μm~4μm。
光輝性顏料的平均縱向長度(lp)與光輝性色調劑的體積平均粒徑(dt)之間的比例(lp/dt)優選為0.017~1.000。
本示例性實施方式中所用的光輝性顏料包含通過以下方式獲得的顏料:在復合顏料技術產品的金屬或金屬化合物層和邊界處的剝離用樹脂層之間的界面處將金屬或金屬化合物層從片狀基材剝離和粉碎,其中所述復合顏料技術產品具有剝離用樹脂層與金屬或金屬化合物層依次層壓在片狀基材上的結構。
用于制備本示例性實施方式中所用光輝性顏料的復合顏料技術產品的金屬或金屬化合物層中使用的金屬或金屬化合物沒有特別限制,只要該金屬或金屬化合物具有展現諸如金屬光澤等功能。然而,使用鋁、銀、金、鎳、鉻、錫、鋅、銦、鈦或銅等,并且使用這些單獨的金屬、金屬化合物、它們的合金及其混合物中的至少一種。在這些實例中,本示例性實施方式中所用的光輝性顏料優選為含有單個金屬的金屬顏料,更優選為鋁顏料。
金屬或金屬化合物層優選通過真空沉積、離子鍍覆或濺射方法形成。金屬或金屬化合物層的厚度沒有特別限制,且優選為30nm~100nm。如果厚度等于或大于30nm,則光輝性顏料的反射性和光輝性被增強。如果厚度等于或小于100nm,則防止了光輝性顏料的表觀比重的增加,并提高了光輝性顏料的分散穩定性。
用于制備本示例性實施方式中所用的光輝性顏料的復合顏料技術產品中的剝離用樹脂層是金屬或金屬化合物層的底涂層,并且是用于增強金屬或金屬化合物層和片狀基材表面之間的剝離性的剝離層。雖然剝離用樹脂層中所用樹脂沒有特別限制,但其優選的實例包括聚乙烯醇、聚乙烯醇縮丁醛、聚乙二醇、聚丙烯酸、聚丙烯酰胺、纖維素衍生物、丙烯酸共聚物或改性尼龍樹脂。
片狀基材被一種樹脂或兩種以上樹脂的混合物的溶液涂布并干燥,從而形成層。該涂布溶液可含有諸如粘度調節劑等添加劑。
剝離用樹脂層通過常用的凹印涂布、輥涂、刮涂、擠出涂布、浸涂或旋涂法等進行涂布。在涂布和干燥之后,根據需要通過壓延處理使表面光滑。
雖然剝離用樹脂層的厚度沒有特別限制,但厚度優選為0.5μm~50μm,更優選為1μm~10μm。如果厚度等于或大于0.5μm,則分散的樹脂量不足。如果厚度等于或小于50μm,則在卷繞復合顏料技術產品的情形中不易出現顏料層界面處的剝離。
雖然用于制備本示例性實施方式中所用的光輝性顏料的復合顏料技術產品中的片狀基材沒有特別限制,但其實例包括聚四氟乙烯、聚乙烯或聚丙烯等的聚烯烴膜;聚對苯二甲酸乙二醇酯的聚酯膜;66尼龍或6尼龍等的聚酰胺膜;和防粘膜,如聚碳酸酯膜、三乙酸酯膜或聚酰亞胺膜。
優選的片狀基材為聚對苯二甲酸乙二醇酯或其共聚物。
雖然片狀基材的厚度沒有特別限制,但厚度優選為10μm~150μm。如果厚度等于或大于10μm,則對于工序中的操作性不存在問題。如果厚度等于或小于150μm,則實現令人滿意的柔韌性,并且沒有卷繞和剝離等方面的問題。
金屬或金屬化合物層可被夾在保護層之間。保護層的實例包括氧化硅層和保護性樹脂層。
雖然氧化硅層沒有特別限制(只要該層含有氧化硅即可),但氧化硅層優選通過溶膠-凝膠方法由諸如四烷氧基硅烷等烷氧基硅或其聚合物形成。
通過對金屬或金屬化合物層涂布溶解有烷氧基硅或其聚合物的醇溶液并將醇溶液加熱和燒制而形成涂布有氧化硅層的膜。
保護性樹脂層中所含的樹脂沒有特別限制。然而,其實例包括聚乙烯醇、聚乙二醇、聚丙烯酸、聚丙烯酰胺或纖維素衍生物,并且優選的實例包括聚乙烯醇或纖維素衍生物。
通過對金屬或金屬化合物層涂布一種樹脂或兩種以上樹脂的混合物的水溶液并將所述水溶液干燥而形成保護性樹脂層。涂布溶液可含有諸如粘度調節劑等添加劑。
氧化硅和樹脂可通過與涂布剝離用樹脂層相同的方法涂布。
雖然保護層的厚度沒有特別限制,但厚度優選為50nm~150nm。如果厚度等于或大于50nm,則獲得保護層的足夠的機械強度。如果厚度等于或小于150nm,則保護層的強度不會變得極其高,并且其不會難以將金屬或金屬化合物層粉碎和分散。另外,不易于發生在保護層和金屬或金屬化合物層界面處的剝離。
可在“保護層”和“金屬或金屬化合物層”之間提供著色材料層。
引入著色材料層以獲得任意的復合著色顏料,并且其沒有特別限制,只要著色材料層可含有能夠施加本示例性實施方式中所用的光輝性顏料的金屬光澤和光輝性之外的任意色調和色相的著色材料即可。作為著色材料層中所用的著色材料,可使用任何染料和顏料。另外,可使用已知染料或已知顏料作為所述染料或顏料。
在此情形中,著色材料層中所用的“顏料”是指顏料化學領域內通常定義的天然顏料、合成有機顏料或合成無機顏料等,并且不同于諸如本示例性實施方式的“復合顏料”等經處理為具有層壓結構的顏料。
雖然形成著色材料層的方法沒有特別限制,但是著色材料層優選通過涂布形成。
在著色材料層中所用著色材料為顏料的情形中,優選的是進一步含有著色材料分散用樹脂,并且優選使用聚乙烯醇縮丁醛或丙烯酸共聚物等作為著色材料分散用樹脂。在此情形中,優選通過以下方式將著色材料層制備為樹脂薄層:將顏料、著色材料分散用樹脂以及必要時的其他添加劑分散或溶解在溶劑中以獲得溶液,將溶液通過旋涂在金屬或金屬化合物層上形成液膜,然后將該液膜干燥。
在用于制備本示例性實施方式中所用的光輝性顏料的復合顏料技術產品的制備中,從操作效率的角度而言,優選的是著色材料層和保護層都通過涂布形成。
制備本示例性實施方式中所用的光輝性顏料的復合顏料技術產品具有分層結構,所述分層結構包含多個剝離用樹脂層和金屬或金屬化合物層依次層壓的結構體。此時,金屬或金屬化合物層層壓的整個多層結構的厚度,即,除片狀基材和緊挨在片狀基材上方的剝離用樹脂層之外的金屬或金屬化合物層-剝離用樹脂層-金屬或金屬化合物層……剝離用樹脂層-金屬或金屬化合物層的厚度優選等于或小于5,000nm。如果該厚度等于或小于5,000nm,則不易出現破裂和剝離,并且即使在將復合顏料技術產品卷成卷形時也實現優異的儲存穩定性。即使在形成顏料的情形中,也實現優異的光輝性,這是優選的。
雖然也示例了剝離用樹脂層和金屬或金屬化合物層依次層壓在片狀基材的兩個表面上的結構,但結構不限于此。
可通過以下方式獲得本示例性實施方式中所用的光輝性顏料:將復合顏料技術產品中的金屬或金屬化合物層在對應于剝離用樹脂層的界面處從片狀基材上剝離,并將金屬或金屬化合物層粉碎和微細化。
雖然剝離處理方法沒有特別限制,但優選進行將復合顏料技術產品浸入液體的方法;或將復合顏料技術產品浸入液體,進行超聲處理、剝離處理和剝離的復合顏料的粉碎處理的方法。
根據上述獲得的光輝性顏料,剝離用樹脂層充當保護性膠體,并且僅通過在溶劑中進行分散處理而容易地獲得穩定的分散液。
將如下詳細描述本示例性實施方式中所用的光輝性顏料的由jisk5906:2009定義的水面擴散面積的測量方法。
通過下述方法從光輝性色調劑中提取光輝性顏料。其后,將提取的樣品用石油精或丙酮洗滌,干燥并粉末成顆粒。水面擴散面積可以如下獲得:將所述顆粒噴灑在水面擴散面積測量設備的水面上并獲得其中被鋁顆粒均勻覆蓋的水面的面積。
從光輝性色調劑中提取光輝性顏料的方法沒有特別限制,并且可通過以下方式分別收集光輝性顏料:將光輝性色調劑放入溶解有粘合劑樹脂的溶劑(例如四氫呋喃)中,將粘合劑樹脂溶解,并通過離心處理使光輝性顏料沉淀。在對光輝性色調劑添加外添劑的情形中,可在將光輝性色調劑放入溶劑之前通過作為預處理的超聲處理將外添劑從色調劑中去除。
光輝性顏料的平均縱向長度是通過以下方式測量的值。
通過將光輝性色調劑定影在諸如紙等記錄介質上并形成光輝性色調劑圖像,使光輝性色調劑中所含光輝性顏料的表面方向沿記錄介質的表面方向對齊。通過使用雙酚a型液體環氧樹脂和固化劑來包埋光輝性色調劑圖像,制備切割用樣品。然后,通過使用利用金剛石刀具的切割機(例如ultracutuct(由leica制造))在100℃將樣品切割成片,并獲得光輝性色調劑圖像的切割面。通過透射電子顯微鏡(tem)觀察光輝性色調劑圖像的切割面,并獲得100個光輝性顏料的縱向長度。通過將放大率設定在可在單個視野中觀察到1~10個光輝性顏料的放大率來進行觀察。從獲得值計算光輝性顏料的算術平均縱向長度,并視之為平均縱向長度。
光輝性顏料的縱向長度是指光輝性顏料的截面的外接圓直徑。
例如,相對于100重量份的色調劑顆粒,光輝性顏料的含量優選為1重量份~50重量份,更優選為15重量份~25重量份。
防粘劑
防粘劑的實例包括:烴蠟;天然蠟,如巴西棕櫚蠟、米蠟或小燭樹蠟;合成、礦物或石油蠟,如褐煤蠟;酯蠟,如脂肪酸酯或褐煤酸酯。防粘劑不限于此。
防粘劑的熔融溫度優選為50℃~110℃,更優選為60℃~100℃。
基于jisk7121-1987“塑料轉變溫度的測試方法”中如何獲得熔融溫度的方法中描述的“熔融峰溫度”由差示掃描量熱法(dsc)得到的dsc曲線來獲得熔融溫度。
例如,相對于全部色調劑顆粒,防粘劑的含量優選為1重量%~20重量%,更優選5重量%~15重量%。
其他添加劑
其他添加劑的實例包括除磁性材料、電荷控制劑、無機粉末和光輝性顏料之外的已知添加劑,例如著色劑。色調劑顆粒含有這些添加劑作為內添劑。
電荷控制劑的實例包括含有季銨鹽化合物、苯胺黑化合物、鋁、鐵或鉻的絡合物的染料,以及三苯甲烷顏料。
作為無機顆粒,可單獨使用一種或組合使用兩種以上已知的無機顆粒,如二氧化硅顆粒、二氧化鈦顆粒、氧化鋁顆粒、二氧化鈰顆粒或者通過將其表面用疏水化劑進行處理而獲得的顆粒。在這些實例中,優選使用具有低于粘合劑樹脂的折射率的二氧化硅顆粒。另外,可對二氧化硅顆粒進行各類表面處理,例如,優選使用表面用例如硅烷偶聯劑、鈦偶聯劑或硅油進行了處理的二氧化硅顆粒。
光輝性顏料之外的著色劑的實例包括已知著色劑,并且根據目標色調進行選擇。作為其他著色劑,可根據需要使用表面經過處理的著色劑,或者著色劑可與分散劑一起使用。
其他著色劑的實例包括各種顏料,例如,炭黑、鉻黃、漢撒黃、聯苯胺黃、陰丹士林黃、喹啉黃、顏料黃、永久橙gtr、吡唑啉酮橙、弗爾肯橙、色淀紅(watchungred)、永久紅、亮洋紅3b、亮洋紅6b、杜邦油紅、吡唑啉酮紅、立索紅、若丹明b色淀、色淀紅c、顏料紅、玫瑰紅、苯胺藍、群青藍、calco油藍、亞甲藍氯化物、酞菁藍、顏料藍、酞菁綠和孔雀綠草酸鹽;或各種染料,例如,吖啶染料、呫噸染料、偶氮染料、苯醌染料、吖嗪染料、蒽醌染料、硫靛染料、二噁嗪染料、噻嗪染料、偶氮甲堿染料、靛藍染料、酞菁染料、苯胺黑染料、聚甲炔染料、三苯甲烷染料、二苯甲烷染料和噻唑染料。
色調劑顆粒的特性
色調劑顆粒可以是具有單層結構的色調劑顆粒,或者可以是具有由芯(芯顆粒)和覆蓋所述芯的覆蓋層(殼層)形成的所謂芯殼結構的色調劑顆粒。
具有芯殼結構的色調劑顆粒優選由以下形成:含有光輝性顏料和粘合劑樹脂以及必要時的其他添加劑(防粘劑)的芯;和含有粘合劑樹脂的覆蓋層。
色調劑顆粒的平均最大厚度c和平均圓當量直徑d
色調劑顆粒具有扁平形狀,并且平均圓當量直徑d優選長于平均最大厚度c。平均最大厚度c與平均圓當量直徑d的比例(c/d)更優選為0.001~0.700,該比例進一步優選為0.100~0.600,并且該比例特別優選為0.300~0.450。
如果該比例(c/d)等于或大于0.001,則保證了色調劑的強度,防止了因圖像形成期間的應力而引起的破裂,防止由顏料暴露導致的帶電減少和由此所致的霧化。另一方面,如果該比例等于或小于0.700,則可實現優異的光輝性。
平均最大厚度c和平均圓當量直徑d通過以下方法測量。
將色調劑顆粒置于扁平表面上,隨后通過對其施加振動而使其無規則地分散。通過以下方式計算平均最大厚度c和平均圓當量直徑d:使用彩色激光顯微鏡“vk-9700”(由keyencecorporation制造)以便將1000個色調劑顆粒放大1000倍,測量從上側觀察時的光輝性色調劑顆粒的最大厚度c和平面的圓當量直徑d,并獲得其算術平均值。
色調劑顆粒截面內的縱軸方向和光輝性顏料的縱軸方向之間的角度
在觀察色調劑顆粒在厚度方向上的截面的情形中,相對于觀察到的全部光輝性顏料,在色調劑顆粒截面的縱軸方向與光輝性顏料的縱軸方向之間角度為-30°~+30°的光輝性顏料的比例(基于數目)等于或大于60%。此外,上述比例進一步優選為70%~95%,特別優選為80%~90%。
如果該比例等于或大于60%,則可實現更優異的光輝性。
在此,將給出色調劑顆粒截面的觀察方法的描述。
利用雙酚a型液體環氧樹脂和固化劑包埋光輝性色調劑圖像,隨后形成切割用樣品。接下來,使用利用金剛石刀具的切割機(例如超微切片機設備(由制造的ultracutuct))以-100°切割所述切割用樣品,并形成觀察用樣品。利用超高分辨率場致發射型掃描電子顯微鏡(由hitachihigh-technologiescorporation制造的s-4800)以可在單個視野中觀察到約1~10個色調劑顆粒的放大率觀察所述觀察用樣品。
具體而言,觀察色調劑顆粒的截面(色調劑顆粒在厚度方向上的截面),利用諸如由mitanicorporation制造的圖像分析軟件(winroof)等圖像分析軟件或者觀察的圖像的打印樣品和分度器,計算觀察的100個色調劑顆粒中在色調劑顆粒截面的縱軸方向與光輝性顏料的縱軸方向之間角度為-30°~+30°的光輝性顏料的件數,并計算其比例。
“色調劑顆粒截面內的縱軸方向”表示與平均圓當量直徑d長于平均最大厚度c的所述色調劑顆粒的厚度方向直交的方向,而“光輝性顏料的縱軸方向”表示光輝性顏料的長度方向。
色調劑的體積平均粒徑優選為3μm~30μm,更優選為5μm~20μm。
色調劑顆粒的體積平均粒徑d50v通過以下方式獲得:通過如multisizerii(由beckmancoulter,inc.制造)等測量設備測得粒徑分布,在基于該粒徑分布劃分的粒徑范圍(區段)內,從較小粒徑側起分別繪制針對體積和數目的累積分布。將對應于16%累積的粒徑定義為具有體積d16v和數目d16p,將對應于50%累積的粒徑定義為具有體積d50v和數目d50p,并將將對應于84%累積的粒徑定義為具有體積d84v和數目d84p。利用所述值將體積粒徑分布指數(gsdv)計算為(d84v/d16v)1/2。
假定色調劑顆粒在厚度方向上的平均長度為1時,在縱軸方向上的平均長度的比例(長徑比)優選為1.5~15,更優選為2~10,進一步優選為3~8。
色調劑顆粒厚度方向上的平均長度和縱軸方向上的平均長度通過以下方式計算:將色調劑顆粒置于光滑表面上,通過施加振動使色調劑顆粒均勻分散,在通過彩色激光顯微鏡“vk-9700”(由keyencecorporation制造)以1,000倍的放大率放大1,000個色調劑顆粒的同時測量從上側觀察的光輝性色調劑顆粒的最大厚度和表面縱軸方向上的長度,并獲得其算術平均值。
外添劑
外添劑的實例包括無機顆粒。無機顆粒的實例包括sio2、tio2、al2o3、cuo、zno、sno2、ceo2、fe2o3、mgo、bao、cao、k2o、na2o、zro2、cao·sio2、k2o·(tio2)n、al2o3·2sio2、caco3、mgco3、baso4和mgso4。
優選用疏水化劑處理作為外添劑的無機顆粒表面。例如,通過將無機顆粒浸在疏水化劑中進行利用疏水化劑的處理。雖然疏水化劑沒有具體限制,其實例包括硅烷偶聯劑、硅油、鈦酸酯偶聯劑和鋁偶聯劑。疏水化劑的一種或兩種以上可單獨或組合使用。
例如,相對于100重量份的無機顆粒,疏水化劑的量一般為1重量份~10重量份。
外添劑的實例還包括樹脂顆粒和(例如聚苯乙烯、聚甲基丙烯酸甲酯(pmma)或三聚氰胺甲醛樹脂等樹脂顆粒)和清潔助劑(例如,高級脂肪酸的金屬鹽,其典型實例包括硬脂酸鋅,以及氟高分子量材料的顆粒)。
例如,相對于色調劑顆粒的量,外添劑的量優選為0.01重量%~5重量%,更優選為0.01重量%~2.0重量%。
色調劑的制備方法
接下來,將給出本示例性實施方式的色調劑的制備方法的描述。
本示例性實施方式的色調劑可以通過以下方式獲得:制備含有光輝性顏料的色調劑顆粒,然后對色調劑顆粒外部添加外添劑。
色調劑顆粒可通過干式制備方法(例如,捏合粉碎法)和濕式制備方法(例如,凝集聚并法、懸浮聚合法或溶解懸浮法)中的任意一種來制備。色調劑顆粒的制備方法并不限于這些制備方法,并且可采用已知的制備方法。
例如,溶解懸浮法是通過以下方式制備和獲得色調劑顆粒的方法:通過將形成色調劑顆粒的原料(如樹脂顆粒和光輝性顏料)溶解或分散在有機溶劑(粘合劑樹脂可溶解在其中)中而獲得溶液,將該溶液分散在含有顆粒分散劑的水溶劑中,之后去除有機溶劑。
另外,凝集聚并法是通過進行以下工序獲得色調劑顆粒的方法:由形成色調劑顆粒的原料(樹脂顆粒和光輝性顏料等)形成凝集體的凝集工序,和將凝集體聚并的聚并工序。
在這些制備方法中,優選的是通過下述溶解懸浮法獲得含有脲改性聚酯樹脂作為粘合劑樹脂的色調劑顆粒。雖然在溶解懸浮法的如下說明中將描述獲得含有防粘劑的色調劑顆粒的方法,色調劑顆粒根據需要含有防粘劑。雖然將描述獲得含有未改性聚酯樹脂和脲改性聚酯樹脂作為粘合劑樹脂的色調劑顆粒的方法,色調劑顆粒可僅含有脲改性聚酯樹脂作為粘合劑樹脂。
制備油相溶液的工序
制備油相溶液,其中將包含未改性聚酯樹脂、具有異氰酸酯基團的聚酯預聚物、胺化合物、光輝性顏料和防粘劑的色調劑顆粒材料溶解或分散在有機溶劑中(制備油相溶液的工序)。制備油相溶液的工序是通過將色調劑顆粒材料溶解或分散在有機溶劑中而獲得色調劑材料混合溶液的工序。
制備油相溶液的方法的實例包括:1)通過將色調劑材料共同溶解或分散在有機溶劑中而制備油相溶液的方法;2)通過預先捏合色調劑材料,然后將捏合的材料溶解或分散在有機溶劑中而制備油相溶液的方法;3)通過將未改性聚酯樹脂、具有異氰酸酯基團的聚酯預聚物和胺化合物溶解在有機溶劑中,然后將光輝性顏料和防粘劑分散在有機溶劑中而制備油相溶液的方法;4)通過將光輝性顏料和防粘劑分散在有機溶劑中,然后將未改性聚酯樹脂、具有異氰酸酯基團的聚酯預聚物和胺化合物溶解在有機溶劑中而制備油相溶液的方法;5)通過將除了具有異氰酸酯基團的聚酯預聚物和胺化合物之外的色調劑顆粒材料(未改性聚酯樹脂、光輝性顏料和防粘劑)溶解或分散在有機溶劑中,然后將具有異氰酸酯基團的聚酯預聚物和胺化合物溶解在有機溶劑中而制備油相溶液的方法;以及6)通過將除了具有異氰酸酯基團的聚酯預聚物或胺化合物之外的色調劑顆粒材料(未改性聚酯樹脂、光輝性顏料和防粘劑)溶解或分散,然后將具有異氰酸酯基團的聚酯預聚物或胺化合物溶解在有機溶劑中而制備油相溶液的方法。制備油相溶液的方法不限于此。
用于油相溶液的有機溶劑的實例包括:酯溶劑,如乙酸甲酯或乙酸乙酯;酮溶劑,如甲基乙基酮或甲基異丙基酮;脂肪族烴溶劑,如己烷或環己烷;以及鹵化烴,如二氯甲烷、氯仿或三氯乙烷。這種有機溶劑優選地溶解粘合劑樹脂,優選地以0重量%~30重量%的比例溶于水,并且優選地具有等于或小于100℃的沸點。在這些有機溶劑中,優選使用乙酸乙酯。
制備懸浮液的工序
接下來,通過將所得油相溶液分散在水相溶液中而制備懸浮液(制備懸浮液的工序)。
隨后,與懸浮液的制備一起,在具有異氰酸酯基團的聚酯預聚物和胺化合物之間引起反應。然后,通過該反應形成脲改性聚酯樹脂。該反應伴隨有分子鏈的交聯反應和伸長反應中的至少一個。具有異氰酸酯基團的聚酯預聚物和胺化合物之間的反應可與去除溶劑的工序一起進行,其將在后面進行描述。
在此,根據聚酯預聚物的異氰酸酯基團和胺化合物之間的反應性選擇反應條件,例如,反應時間優選為10分鐘~40小時,并優選為2小時~24小時。反應溫度優選為0℃~150℃,并優選為40℃~98℃。為了形成脲改性聚酯樹脂,可根據需要使用已知催化劑(月桂酸二丁基錫或月桂酸二辛基錫等)。換言之,可對油相溶液或懸浮液添加催化劑。
水相溶液的實例包括通過將諸如樹脂顆粒分散劑或無機顆粒分散劑等顆粒分散劑分散在水性溶劑中而獲得的水相溶液。水相溶液的實例還包括通過將顆粒分散劑分散在水性溶液并將高分子分散劑溶解而獲得的水相溶液。另外,可對水相溶液添加已知添加劑,如表面活性劑。
水性溶液的實例包括水(例如,一般而言,離子交換水、蒸餾水或純水)。水性溶液可以是連同水一起含有有機溶劑的溶劑,所述有機溶劑例如為醇(如甲醇、異丙醇或乙二醇)、二甲基甲酰胺、四氫呋喃、溶纖劑(如甲基溶纖劑)或低級酮(如丙酮或甲基乙基酮)。
有機顆粒分散劑的實例包括親水性有機顆粒分散劑。有機顆粒分散劑的實例包括聚(甲基)丙烯酸烷基酯樹脂(如聚甲基丙烯酸甲酯)、聚苯乙烯樹脂和聚(苯乙烯丙烯腈)樹脂的顆粒。
無機分散劑的實例包括親水性無機顆粒分散劑。無機顆粒分散劑的具體實例包括二氧化硅、氧化鋁、氧化鈦、碳酸鈣、碳酸鎂、磷酸三鈣、粘土、硅藻土或膨潤土的顆粒,優選使用碳酸鈣的顆粒。可以單獨使用一種或者組合使用兩種以上無機顆粒分散劑。
可用具有羧基的聚合物處理顆粒分散劑的表面。
具有羧基的聚合物的實例包括選自鹽(例如堿金屬鹽、堿土金屬鹽、銨鹽和胺鹽)中的至少一種和α,β-單烯鍵式不飽和羧酸酯的共聚物,所述鹽通過如下方式獲得:用堿金屬、堿土金屬、氨或胺等中和α,β-單烯鍵式不飽和羧酸或α,β-單烯鍵式不飽和羧酸中的羧基。具有羧基的聚合物的實例還包括通過以下方式獲得的鹽(例如堿金屬鹽、堿土金屬鹽、銨鹽和胺鹽):用堿金屬、堿土金屬、氨或胺等中和α,β-單烯鍵式不飽和羧酸和α,β-單烯鍵式不飽和羧酸酯的共聚物中的羧基。具有羧基的聚合物可以單獨使用一種,或組合使用兩種以上。
α,β-單烯鍵式不飽和羧酸的代表性實例包括α,β-不飽和單羧酸(如丙烯酸、甲基丙烯酸或巴豆酸)和α,β-不飽和二羧酸(如馬來酸、富馬酸或衣康酸)。α,β-單烯鍵式不飽和羧酸酯的代表性實例包括(甲基)丙烯酸的烷基酯、具有烷氧基的(甲基)丙烯酸酯、具有環己基的(甲基)丙烯酸酯、具有羥基的(甲基)丙烯酸酯和聚亞烷基二醇單(甲基)丙烯酸酯。
高分子分散劑的實例包括親水性高分子分散劑。高分子分散劑的具體實例包括具有羧基且不具有疏水性基團(例如羥基丙氧基或甲氧基)的高分子分散劑(水溶性纖維素醚,例如羧甲基纖維素或羧乙基纖維素)。
去除溶劑的工序
接下來,通過從獲得的懸浮液中去除有機溶劑而獲得色調劑顆粒分散液(去除溶劑的工序)。去除溶劑的工序是通過去除分散在懸浮液中的水相溶液的液滴中所含的有機溶劑而制備色調劑顆粒的工序。從懸浮液去除有機溶劑可在制備懸浮液的工序之后馬上進行,或者可在制備懸浮液的工序完成1分鐘以上之后進行。
在去除溶劑的過程中,例如,優選通過將所得懸浮液冷卻或加熱至0℃~100℃來從懸浮液中去除有機溶劑。
作為去除有機溶劑的具體方法,示例了如下方法。
(1)對懸浮液吹送氣流并且將懸浮液表面上的氣相強制更新的方法。在此情形中,可將氣體吹送到懸浮液中。
(2)降低壓力的方法。在此情形中,可通過氣體的充填將懸浮液表面上的氣相強制更新,或者可將氣體進一步吹送到懸浮液中。
通過所述工序獲得色調劑顆粒。
在此,在去除溶劑的工序結束之后,獲得在對色調劑顆粒分散液中形成的色調劑顆粒進行了已知清潔工序、固液分離工序和干燥工序之后的干燥狀態的色調劑顆粒。
從帶電性出發,在清潔工序中,優選的是通過離子交換水充分進行置換清潔。
雖然沒有特別限制,但從生產率出發,在固液分離工序中,優選的是進行抽濾或壓濾等。雖然沒有特別限制,但從生產率出發,在干燥工序中,優選的是進行冷凍干燥、閃噴干燥、流化干燥或振動型流化干燥等。
本示例性實施方式的色調劑通過例如對所得干燥狀態的色調劑顆粒添加外添劑并將色調劑顆粒與外添劑混合來制造。
混合優選用v型共混器、亨舍爾混合機或lodige混合機等來進行。
此外,根據需要,可利用振動分級器或風力分級器等除去色調劑的粗大顆粒。
在本示例性實施方式中,可以使用凝集聚并法,其中,容易控制色調劑顆粒的形狀和粒徑,并且色調劑顆粒結構(如芯殼結構)可在較寬范圍內控制。下文中,將給出通過凝集聚并法制備色調劑顆粒的方法的詳細描述。
本示例性實施方式的凝集聚并法包括通過將形成色調劑顆粒的原料分散而形成樹脂顆粒(乳化顆粒)的分散工序、形成樹脂顆粒的凝集體的凝集工序和使凝集體聚并的聚并步驟。
分散工序
樹脂顆粒分散液可通過常見聚合方法((如乳化聚合法、懸浮聚合法或分散聚合法)制造,或者可通過對利用分散機將水性介質和粘合劑樹脂混合而獲得的溶液施加剪切力來制造。此時,可通過加熱使得樹脂成分具有低粘度,從而形成顆粒。另外,可以使用分散劑來使分散的樹脂顆粒穩定化。只要樹脂是油性并且溶于對水的溶解度相對較低的溶劑,則通過以下方式制備樹脂顆粒分散液:將樹脂溶解在溶劑中,將顆粒連同分散劑和聚合物電解質一起分散于水中,然后通過加熱或減壓來蒸發溶劑。
水性介質的實例包括水(如蒸餾水或離子交換水)和醇。優選使用水。
乳化工序中使用的分散劑的實例包括:水溶性聚合物,如聚乙烯醇、甲基纖維素、乙基纖維素、羥乙基纖維素、羧甲基纖維素、聚丙烯酸鈉或聚甲基丙烯酸鈉;表面活性劑,如陰離子型表面活性劑(例如,十二烷基苯磺酸鈉、十八烷基硫酸鈉、油酸鈉、月桂酸鈉或硬脂酸鉀)、陽離子型表面活性劑(例如,月桂基胺乙酸鹽、硬脂基胺乙酸鹽或月桂基三甲基氯化銨)、兩性離子型表面活性劑(例如,月桂基二甲基氧化胺)或非離子型表面活性劑(例如,聚氧乙烯烷基醚、聚氧乙烯烷基苯基醚、聚氧乙烯烷基胺);或無機鹽,如(磷酸三鈣、氫氧化鋁、硫酸鈣、碳酸鈣或碳酸鋇)。
用于制備乳化液的分散機的實例包括均質器、均相混合機、壓力捏合機、擠出機和介質分散機。至于樹脂顆粒的尺寸,平均粒徑(體積平均粒徑)優選等于或小于1.0μm,優選為60nm~300nm,進一步優選為150nm~250nm。如果平均粒徑等于或大于60nm,則存在樹脂顆粒溶劑凝集的情形,因為樹脂顆粒在分散液中易于不穩定。如果平均粒徑等于或小于1.0μm,則存在色調劑的粒徑分布變窄的情形。
為了制備防粘劑分散液,將防粘劑與離子型表面活性劑或聚合物電解質(如聚合物酸或聚合物堿)一起分散在水中,以等于或大于防粘劑熔融溫度的溫度加熱所得物,并且利用施加高剪切力的均質器或壓力排出型分散機進行分散處理。通過這樣的處理,獲得防粘劑分散液。在分散處理中,可以向分散液中添加如聚氯化鋁等無機化合物。優選的無機化合物的實例包括聚氯化鋁、硫酸鋁、高堿性聚氯化鋁(bac)、聚氫氧化鋁和氯化鋁。在這些實例中,優選使用聚氯化鋁和硫酸鋁等。
通過分散處理,獲得含有體積平均粒徑等于或小于1μm的防粘劑顆粒的防粘劑分散液。防粘劑顆粒的體積平均粒徑更優選為100nm~500nm。
如果體積平均粒徑等于或大于100nm,防粘劑成分一般容易進入色調劑中,其受到使用的粘合劑樹脂的特性影響。如果體積平均粒徑等于或小于500nm,則實現了防粘劑在色調劑中的令人滿意的分散狀態。
為了制備光輝性顏料分散液,可以使用已知的分散方法,并且可以使用如旋轉剪切型均質器、具有介質的球磨機、砂磨機、戴諾磨機或超細磨機(ultimizer)等典型分散單元,而且沒有限制。光輝性顏料連同離子型表面活性劑和聚合物電解質(如聚合物酸或聚合物堿)一起分散在水中。只要尺寸等于或小于20μm,就可容許分散的金屬顏料的體積平均粒徑。然而,該體積平均粒徑優選為3μm~16μm,這是因為該體積平均粒徑不破壞凝集性并且可實現光輝性顏料在色調劑中的令人滿意的分散。
可通過以下方式制備被粘合劑樹脂覆蓋的光輝性顏料的分散液:將光輝性顏料和粘合劑樹脂分散或溶解和混合在溶劑中,并通過相轉化乳化或剪切乳化將所得物分散在水中。
凝集工序
在凝集工序中,通過將樹脂顆粒分散液、光輝性顏料分散液和防粘劑分散液等混合而獲得混合物,并以等于或小于樹脂顆粒的玻璃化轉變溫度的溫度加熱混合溶液以便凝集,形成凝集顆粒。在許多情形中,在攪拌的同時將混合溶液的ph控制在酸性,從而形成凝集顆粒。在所述攪拌條件下,可以將比例(c/d)設定在優選范圍內。更具體而言,在凝集顆粒形成階段,通過高速攪拌混合溶液并加熱混合溶液可以使比例(c/d)減小,并且通過在更低溫度下以降低的速度攪拌混合溶液可以使比例(c/d)增大。另外,ph優選為2~7,此時也可以有效地使用凝集劑。
在凝集工序中,防粘劑可被添加并與各種分散液(如樹脂顆粒分散液)一次性混合在一起,或者可被分開和多次添加。
作為凝集劑,優選使用極性與用于分散劑的表面活性劑的極性相反的表面活性劑、無機金屬鹽以及二價以上的金屬絡合物。由于可以減少表面活性劑的量并增強帶電特性,特別優選使用金屬絡合物。
作為無機金屬鹽,特別優選使用鋁鹽及其聚合物。至于無機金屬鹽的價態,二價無機金屬鹽比一價無機金屬鹽更適合,三價無機金屬鹽比二價無機金屬鹽更適合,四價無機金屬鹽比三價無機金屬鹽更適合,并且在相同價態的情形中,更適合的是聚合型無機金屬鹽聚合物,以便獲得較窄的粒徑分布。
在本示例性實施方式中,優選使用含鋁的四價無機金屬鹽的聚合物以獲得較窄的粒徑分布。
當獲得凝集顆粒的所需粒徑時,可通過額外添加樹脂顆粒而制備具有芯材凝集顆粒的表面被樹脂覆蓋的構造的色調劑(覆蓋工序)。從帶電性和顯影性出發此情形中的構造是優選的,這是因為防粘劑和光輝性顏料不易暴露于色調劑的表面。在額外添加樹脂顆粒分散液的情形中,可添加凝集劑或在額外添加之前進行ph調節。
聚并工序
在聚并工序中,通過以下方式使凝集顆粒聚并:通過在凝集工序中的攪拌條件下將凝集顆粒懸浮液的ph增加至3~9來停止凝集的進行,并以等于或高于樹脂的玻璃化轉變溫度的溫度進行加熱。
在用樹脂覆蓋芯材、凝集顆粒的情形中,樹脂也聚并以覆蓋芯材、凝集顆粒。可容許任何加熱時間,只要聚并可實現,并且聚并可在一段約0.5小時~約10小時的時間內進行。
在聚并后將所得物冷卻,并且獲得聚并顆粒。在冷卻的過程中,可通過在樹脂的玻璃化轉變溫度附近(玻璃化轉變溫度±10℃的范圍內)降低冷卻速率,即進行緩慢冷卻,來促進結晶。
對通過聚并獲得的聚并顆粒進行如過濾等固液分離工序和必要時的清潔工序和干燥工序,由此獲得色調劑顆粒。
出于電荷調節、施加流動性和施加電荷交換性等的目的,無機氧化物等(其代表性實例包括二氧化硅、二氧化鈦和氧化鋁)可作為外添劑添加并粘附至所得色調劑顆粒。優選的外部添加方法和優選的外添劑量如上所述。
除了所述無機氧化物等以外,還可以添加如電荷控制劑、有機顆粒、潤滑劑和研磨劑等其他成分(顆粒)作為外添劑。
雖然電荷控制劑沒有特別限制,但優選使用無色或淺色的電荷控制劑。其實例包括季銨鹽化合物、苯胺黑化合物、鋁或鉻的絡合物以及三苯甲烷類顏料。
有機顆粒的實例包括乙烯基樹脂、聚酯樹脂或硅酮樹脂等通常用作添加至色調劑表面的外添劑的顆粒。另外,無機顆粒和有機顆粒用作流動助劑或清潔助劑等。
潤滑劑的實例包括脂肪酸酰胺,如亞乙基二硬脂酸酰胺或油酸酰胺;和脂肪酸金屬鹽,如硬脂酸鋅和硬脂酸鈣。
研磨劑的實例包括如上所述的二氧化硅、氧化鋁和氧化鈰。
靜電荷圖像顯影劑
本示例性實施方式的靜電荷圖像顯影劑至少含有本示例性實施方式的色調劑。本示例性實施方式的靜電荷圖像顯影劑可以是僅包含本示例性實施方式的色調劑的單組分顯影劑,也可以是色調劑與載體混合的雙組分顯影劑。
載體沒有特別限制,例舉已知載體。載體的實例包括由磁性顆粒制成的芯材的表面被樹脂覆蓋的覆蓋型載體;磁性顆粒分散并共混在基質樹脂中的磁性顆粒分散型載體;以及多孔磁性顆粒中浸漬有樹脂的樹脂浸漬型載體。
磁性顆粒分散型載體和樹脂浸漬型載體可以是所述載體的構成顆粒形成芯材并且其表面被樹脂覆蓋的載體。
磁性粉末的實例包括:磁性金屬,如氧化鐵、鎳或鈷;以及磁性氧化物,如鐵氧體和磁鐵礦。
覆蓋用樹脂和基質樹脂的實例包括聚乙烯、聚丙烯、聚苯乙烯、聚乙酸乙烯酯、聚乙烯醇、聚乙烯縮丁醛、聚氯乙烯、聚乙烯基醚、聚乙烯基酮、氯乙烯-乙酸乙烯酯共聚物、苯乙烯-丙烯酸共聚物、或含有有機硅氧烷鍵的直鏈硅酮樹脂或其改性物質;氟樹脂;聚酯;聚碳酸酯;酚醛樹脂;和環氧樹脂。覆蓋用樹脂和基質樹脂可含有諸如導電性顆粒等添加劑。
導電性顆粒的實例包括:金屬,例如金、銀和銅;以及炭黑、二氧化鈦、氧化鋅、氧化錫、硫酸鋇、硼酸鋁或鈦酸鉀等顆粒。
為了覆蓋芯材的表面,例舉利用覆蓋層形成用溶液的方法,所述溶液通過將覆蓋用樹脂和各種添加劑(按需使用)溶解在適當溶劑中而獲得。所述溶劑沒有特別限制,并且考慮所用樹脂的類型和施加適用性等來進行選擇。樹脂覆蓋方法的具體實例包括:將芯材浸漬在覆蓋層形成用溶液中的浸漬法;將覆蓋層形成用溶液噴至芯材的表面上的噴霧法;以使芯材通過流動空氣而漂浮的狀態噴灑覆蓋層形成用溶液的流化床法;以及使載體的芯材與覆蓋層形成用溶液在捏合涂布機中混合然后除去溶劑的捏合涂布機法。
雙組分顯影劑中色調劑與載體的混合比(重量比)優選為色調劑:載體=1:100~30:100,更優選為3:100~20:100。
圖像形成設備/圖像形成方法
將給出本示例性實施方式的圖像形成設備和圖像形成方法的描述。
本示例性實施方式的圖像形成設備包含圖像保持部件;對圖像保持部件的表面充電的充電單元;在圖像保持部件的經充電表面上形成靜電荷圖像的靜電荷圖像形成單元;容納靜電荷圖像顯影劑并利用靜電荷圖像顯影劑使在所述圖像保持部件的表面上形成的所述靜電荷圖像顯影為色調劑圖像的顯影單元;將在所述圖像保持部件的表面上形成的所述色調劑圖像轉印至記錄介質的表面上的轉印單元;和將轉印到所述記錄介質表面上的所述色調劑圖像定影的定影單元。
本示例性實施方式的靜電荷圖像顯影劑作為所述靜電荷圖像顯影劑施加。
本示例性實施方式的圖像形成設備執行圖像形成方法(本示例性實施方式的圖像形成方法),所述圖像形成方法包括:對圖像保持部件的表面充電的充電工序;在所述圖像保持部件的經充電表面上形成靜電荷圖像的靜電荷形成工序;通過本示例性實施方式的靜電荷圖像顯影劑將在所述圖像保持部件表面上形成的靜電荷圖像顯影為色調劑圖像的顯影工序;將在圖像保持部件的表面上形成的所述色調劑圖像轉印至記錄介質的表面上的轉印工序;和將轉印到記錄介質的表面上的色調劑圖像定影的定影工序。
作為本示例性實施方式的圖像形成設備,已知圖像形成設備,例如:將圖像保持部件表面上形成的色調劑圖像直接轉印至記錄介質的直接轉印型設備;將圖像保持部件表面上形成的色調劑圖像一次轉印至中間轉印部件的表面上,然后將轉印至中間轉印部件表面上的色調劑圖像二次轉印至記錄介質表面的中間轉印型設備;設置有清潔單元的設備,該清潔單元用于在充電前且轉印色調劑圖像后清潔圖像保持部件的表面;或者設置有除電單元的設備,該設備在施加充電之前和轉印色調劑圖像之后通過用除電光照射圖像保持部件的表面而進行除電。
在中間轉印型設備的情形中,例如,對轉印單元施用一種結構,所述結構包含其表面上轉印有色調劑圖像的中間轉印部件、將在圖像保持部件表面上形成的色調劑圖像一次轉印至中間轉印部件表面上的一次轉印單元、和將轉印至中間轉印部件表面上的色調劑圖像二次轉印至記錄介質表面上的二次轉印單元。
在本示例性實施方式的圖像形成設備中,例如,包含顯影單元的部分可具有能夠從圖像形成設備上拆卸的盒式結構(處理盒)。作為所述處理盒,例如,優選使用容納本示例性實施方式的靜電荷圖像顯影劑且設置有顯影單元的處理盒。
下文中將給出本示例性實施方式的圖像形成設備的實例的描述。然而,所述圖像形成設備并不限于此。將描述附圖中示出的主要部件,而省略其他部件的描述。
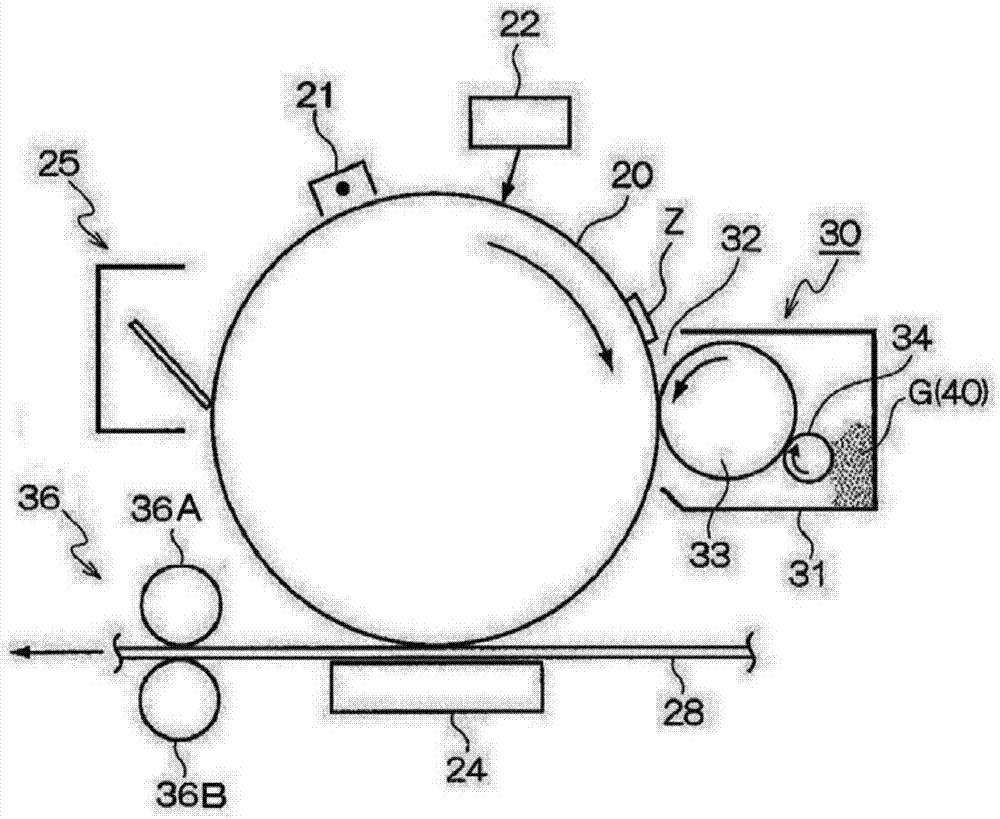
圖2是示意性圖解本示例性實施方式的示例性圖像形成設備的構造圖,所述圖像形成設備包含施加有本示例性實施方式的靜電荷圖像顯影劑的顯影裝置。
在圖中,本示例性實施方式的圖像形成裝置包括沿預定方向旋轉的作為圖像保持部件的感光鼓20。在感光鼓20周圍依次設置有:充電裝置21,其對感光鼓20充電;曝光裝置22,其例如作為在感光鼓20上形成靜電荷圖像z的靜電荷圖像形成裝置;顯影裝置30,其使在感光鼓20上形成的靜電荷圖像z顯影為可視圖像;轉印裝置24,其將感光鼓20上可視化的色調劑圖像轉印至作為記錄介質的記錄紙28;和清潔裝置25,其清除感光鼓20上殘留的色調劑。
如圖2所示,在本示例性實施方式中,顯影裝置30具有容納包含色調劑40的顯影劑g的顯影外殼31。顯影開口32在顯影外殼31中打開以面向感光鼓20。作為色調劑保持體的顯影輥(顯影電極)33被設置為面向顯影開口32。通過對顯影輥33施加預定顯影偏壓,在夾在感光鼓20與顯影輥33之間的區域(顯影區)中形成顯影電場。此外,在顯影外殼31中提供作為電荷注入部件的電荷注入輥(注入電極)34以便面向顯影輥33。具體而言,在本示例性實施方式中,電荷注入輥34還充當對顯影輥33供給色調劑40的色調劑供給輥。
此處,可選擇電荷注入輥34的旋轉方向。不過,考慮到色調劑供給性質和電荷注入特性,電荷注入輥34優選在與顯影輥33相對的部分沿與顯影輥33相同的方向并在有圓周速度差(例如,1.5倍以上)的條件下旋轉,將色調劑40夾在電荷注入輥34和顯影輥33之間的區域,并在刮擦的同時注入電荷。
接下來將給出本示例性實施方式的圖像形成裝置的運行的描述。
如果圖像形成過程開始,則充電裝置21首先對感光鼓20的表面充電,曝光裝置22將靜電荷圖像z寫在經充電的感光鼓20上,而顯影裝置30使靜電荷圖像z顯影為色調劑圖像(其為可視圖像)。其后,將感光鼓20上的色調劑圖像輸送至轉印部分,并且轉印裝置24將在感光鼓20上的色調劑圖像靜電轉印至作為記錄介質的記錄紙28。清潔裝置25清潔在感光鼓20上殘余的色調劑。其后,設置有定影部件36a(定影帶和定影輥等)和加壓部件36b的定影裝置36將記錄紙28上的色調劑圖像定影,獲得圖像。
處理盒/色調劑盒
將給出本示例性實施方式的處理盒的描述。
本示例性實施方式的處理盒是如下處理盒,其包括容納本示例性實施方式的靜電荷圖像顯影劑并通過該靜電荷圖像顯影劑使在圖像保持部件表面上形成的靜電荷圖像顯影形為色調劑圖像的顯影單元,并且能夠從圖像形成設備上拆卸。
本示例性實施方式的處理盒并不限于所述構造,并且可具有如下構造,其包含顯影裝置,且必要時包含選自諸如圖像保持部件、充電單元、靜電荷圖像形成單元和轉印單元等其他單元中的至少一個。
雖然將在下面描述本示例性實施方式的處理盒的實例,但處理盒并不限于此。另外,將描述附圖中示出的主要部件,而省略其他部件的描述。
圖3是示意性圖解本示例性實施方式的處理盒的構造圖。
圖3所示的處理盒200在設置有接軌116和曝光用開口118的外殼117內一體化地組成和保持例如感光體107(圖像保持部件的實例)、在感光體107的周圍設置的充電輥108(充電單元的實例)、顯影裝置111(顯影單元的實例)、以及清潔裝置113(清潔單元的實例),由此構成處理盒。
在圖3中,109表示曝光裝置(靜電荷圖像形成單元的實例),112表示轉印裝置(轉印單元的實例),115表示定影裝置(定影單元的實例),而300表示記錄紙(記錄介質的實例)。
接下來,將給出本示例性實施方式的色調劑盒的描述。
本示例性實施方式的色調劑盒可容納本示例性實施方式的色調劑,并能夠從圖像形成裝置上拆卸。本示例性實施方式的色調劑盒僅必需至少容納色調劑,并且,例如,顯影劑可根據圖像形成裝置的機制而被容納在其中。本示例性實施方式的色調劑盒可具有含有本示例性實施方式的色調劑的容器。
圖2所示的圖像形成設備是具有如下構造的圖像形成設備,其中色調劑盒(未示出)可自由拆卸,并且顯影裝置30通過未在圖中示出的色調劑供給管與色調劑盒連接。在色調劑盒中所容納的色調劑的量變少時,可更換色調劑盒。
實施例
雖然下面將提供參照實施例對本示例性實施方式的詳細描述,但本示例性實施方式不限于這些實施例。在以下描述中,除非另作說明,所有“份”和“%”的描述都是基于重量。
未改性聚酯樹脂(1)的制備
·對苯二甲酸:1243份
·雙酚a的氧化乙烯加合物:1830份
·雙酚a的氧化丙烯加合物:840份
在于180℃加熱的同時將上述成分混合,隨后向其中加入3份二丁基氧化錫,在于220℃加熱混合物的同時將水蒸發,并由此獲得不飽和聚酯樹脂。所得不飽和聚酯樹脂的玻璃化轉變溫度tg為60℃,酸值為3mgkoh/g,且羥基值為1mgkoh/g。
聚酯預聚物(1)的制備
·對苯二甲酸:1243份
·雙酚a的氧化乙烯加合物:1830份
·雙酚a的氧化丙烯加合物:840份
在于180℃加熱的同時將上述成分混合,隨后向其中加入3份二丁基氧化錫,在于220℃加熱混合物的同時將水蒸發,并獲得聚酯。將350份所得聚酯、50份甲代亞苯基二異氰酸酯和450份乙酸乙酯倒入容器中,將其混合物在130℃加熱3小時,并由此獲得具有異氰酸酯基的聚酯預聚物(1)(下文中稱作異氰酸酯改性聚酯預聚物(1))。
酮亞胺化合物(1)的制備
將50份甲基乙基酮和150份六亞甲基二胺倒入容器中,并將混合物在60℃攪拌,由此獲得酮亞胺化合物(1)。
光輝性顏料分散液(1)的制備
(a)通過旋涂法用具有下述組成的樹脂層涂布液涂布膜厚度為100μm的pet膜而制備均勻液膜,隨后將該液膜干燥,由此制備樹脂薄膜層。
(樹脂層涂布液)
·聚乙烯醇縮丁醛樹脂(s-lecbl-10,由sekisuichemicalco.,ltd.制造):3.0%
·甘油:2.0%
·異丙醇(ipa):殘留量
(b)使用由vacuumdevice制造的ve-1010真空沉積設備,以便在上述剝離用樹脂層上形成膜厚度為100μm的鋁沉積層。
(c)利用asonecorporation制造的超聲清潔器(vs-150)在ipa中進行6.5小時的分散處理,以便剝離并微細化通過所述方法形成的pet膜,所述pet膜具有包含剝離用樹脂層和鋁沉積層的層壓結構,由此制備光輝性顏料分散液(1)。
通過所述方法獲得的光輝性顏料分散液中的顏料含量為5.0重量%。
光輝性顏料分散液(1)中所含光輝性顏料的水面擴散面積和平均縱向長度將在表1中示出。
光輝性顏料分散液(2)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(2),不同之處在于,在制備光輝性顏料分散液(1)的(c)中進行25小時的微細化分散處理。
光輝性顏料分散液(3)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(3),不同之處在于,在制備光輝性顏料分散液(1)的(c)中進行6小時的微細化分散處理。
光輝性顏料分散液(4)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(4),不同之處在于,在制備光輝性顏料分散液(1)的(c)中進行2.5小時的微細化分散處理。
光輝性顏料分散液(5)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(5),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為60nm的鋁沉積層。
光輝性顏料分散液(6)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(6),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為60nm的鋁沉積層,并且在制備其的(c)中進行25小時的微細化分散處理。
光輝性顏料分散液(7)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(7),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為60nm的鋁沉積層,并且在制備其的(c)中進行4小時的微細化分散處理。
光輝性顏料分散液(8)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(8),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為60nm的鋁沉積層,并且在制備其的(c)中進行3小時的微細化分散處理。
光輝性顏料分散液(9)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(9),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為20nm的鋁沉積層。
光輝性顏料分散液(10)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(10),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為20nm的鋁沉積層,并且在制備其的(c)中進行25小時的微細化分散處理。
光輝性顏料分散液(11)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(11),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為20nm的鋁沉積層,并且在制備其的(c)中進行4小時的微細化分散處理。
光輝性顏料分散液(12)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(12),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為20nm的鋁沉積層,并且在制備其的(c)中進行3小時的微細化分散處理。
光輝性顏料分散液(13)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(13),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為90nm的鋁沉積層。
光輝性顏料分散液(14)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(14),不同之處在于,在制備光輝性顏料分散液(1)的(b)中形成膜厚度為15nm的鋁沉積層。
光輝性顏料分散液(15)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(15),不同之處在于,在制備光輝性顏料分散液(1)的(c)中進行63小時的微細化分散處理。
光輝性顏料分散液(16)的制備
以與制備光輝性顏料分散液(1)相同的方式制備光輝性顏料分散液(16),不同之處在于,在制備光輝性顏料分散液(1)的(c)中進行2.0小時的微細化分散處理。
防粘劑分散液(1)的制備
·石蠟(熔融溫度:89℃):30份
·乙酸乙酯:270份
以在10℃冷卻的狀態通過微型珠分散器(dcp磨機)將上述成分濕式粉碎,獲得防粘劑分散液(1)。
油相溶液(1)的制備
·未改性聚酯樹脂(1):136份
·光輝性顏料分散液(1):500份
·乙酸乙酯:56份
將上述成分攪拌和混合,然后向所得混合物中添加75份防粘劑分散液(1),攪拌混合物,獲得油相溶液(1)。
苯乙烯丙烯酸樹脂顆粒分散液(1)的制備
·苯乙烯:370份
·丙烯酸正丁酯:30份
·丙烯酸:4份
·十二烷硫醇:24份
·四溴化碳:4份
將通過混合和溶解上述成分而獲得的混合物放入水溶液中,在燒瓶中乳化,所述水溶液通過將6份非離子型表面活性劑(由sanyochemicalindustries,ltd.制造的nonipol400)和10份陰離子型表面活性劑(由dskco.,ltd.制造的neogensc)溶解在560份離子交換水中獲得,將通過使4份過硫酸銨溶解在50份離子交換水中獲得的水溶液倒入混合物中同時使混合物混合10分鐘,對混合物進行氮吹掃,隨后攪拌燒瓶中內含物的同時在油浴中加熱直至內含物的溫度達到70℃,并且使乳化聚合如此繼續5小時。因此,獲得苯乙烯丙烯酸樹脂顆粒分散液(1),其中分散有平均粒徑為180nm且重均分子量(mw)為15,500的樹脂顆粒。苯乙烯丙烯酸樹脂顆粒的玻璃化轉變溫度為59℃。
水相溶液(1)的制備
·苯乙烯丙烯酸樹脂顆粒分散液(1):60份
·celogenbs-h(dskco.,ltd.)的2%水溶液:200份
·離子交換水:200份
將上述成分攪拌并混合,由此獲得水相溶液(1)。
實施例1
色調劑顆粒(1)的制備
·油相溶液(1):300份
·異氰酸酯改性聚酯預聚物(1):25份
·酮亞胺化合物(1):0.5份
將上述成分放入容器中,通過用均質器(ika制造的ultra-turrax)攪拌所述成分2分鐘而獲得油相溶液(1p),隨后將1000份水相溶液(1)加入容器中,并通過均質器將混合物攪拌20分鐘。隨后,通過推進型攪拌器在常壓(1atm)將混合物溶液在室溫(25℃)下攪拌48小時,通過引起異氰酸酯改性聚酯預聚物(1)和酮亞胺化合物(1)之間的反應而制備脲改性聚酯樹脂,去除有機溶劑,獲得粒狀物。然后,將粒狀物用水洗滌,干燥并分級,由此獲得色調劑顆粒(1)。色調劑顆粒的體積平均粒徑為7.6μm,且長徑比為3.3。
光輝性色調劑(1)的制備
通過樣品磨機以10000rpm將100份色調劑顆粒(1)、1.5份疏水性二氧化硅(由nipponaerosilco.,ltd.制造的ry50)和1.0份疏水性二氧化鈦(由nipponaerosilco.,ltd.制造的t805)混合30秒。其后,通過具有45μm的篩網的振動分級器將混合物分級,由此獲得光輝性色調劑(1)。
實施例2
以與制備油相溶液(1)相同的方式獲得油相溶液(2),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(2)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(2),不同之處在于使用油相溶液(2)。
實施例3
以與制備油相溶液(1)相同的方式獲得油相溶液(3),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(3)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(3),不同之處在于使用油相溶液(3)。
實施例4
以與制備油相溶液(1)相同的方式獲得油相溶液(4),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(4)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(4),不同之處在于使用油相溶液(4)。
實施例5
以與制備油相溶液(1)相同的方式獲得油相溶液(5),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(5)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(5),不同之處在于使用油相溶液(5)。
實施例6
以與制備油相溶液(1)相同的方式獲得油相溶液(6),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(6)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(6),不同之處在于使用油相溶液(7)。
實施例7
以與制備油相溶液(1)相同的方式獲得油相溶液(7),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(7)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(7),不同之處在于使用油相溶液(7)。
實施例8
以與制備油相溶液(1)相同的方式獲得油相溶液(8),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(8)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(8),不同之處在于使用油相溶液(8)。
實施例9
以與制備油相溶液(1)相同的方式獲得油相溶液(9),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(9)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(9),不同之處在于使用油相溶液(9)。
實施例10
以與制備油相溶液(1)相同的方式獲得油相溶液(10),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(10)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(10),不同之處在于使用油相溶液(10)。
實施例11
以與制備油相溶液(1)相同的方式獲得油相溶液(11),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(11)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(11),不同之處在于使用油相溶液(11)。
實施例12
以與制備油相溶液(1)相同的方式獲得油相溶液(12),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(12)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(12),不同之處在于使用油相溶液(12)。
比較例1
以與制備油相溶液(1)相同的方式獲得油相溶液(13),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(13)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(13),不同之處在于使用油相溶液(13)。
比較例2
以與制備油相溶液(1)相同的方式獲得油相溶液(14),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(14)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(14),不同之處在于使用油相溶液(14)。
比較例3
以與制備油相溶液(1)相同的方式獲得油相溶液(15),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(15)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(15),不同之處在于使用油相溶液(15)。
比較例4
以與制備油相溶液(1)相同的方式獲得油相溶液(16),不同之處在于在油相溶液(1)的制備中使用光輝性顏料分散液(16)代替光輝性顏料分散液(1)。
另外,以與制備光輝性色調劑(1)相同的方式獲得光輝性色調劑(16),不同之處在于使用油相溶液(16)。
實施例13
樹脂顆粒水分散液(1)的制備
·對苯二甲酸:30摩爾份
·富馬酸:70摩爾份
·雙酚a的氧化乙烯加合物:5摩爾份
·雙酚a的氧化丙烯加合物:95摩爾份
在具有5升含量且設置有攪拌器、氮引入管、溫度傳感器和整流器的燒瓶中制備上述材料,在1小時內將溫度升高至220℃,并將1份四乙氧基鈦放入100份上述材料中。在0.5小時內使溫度升高至230℃同時蒸發生成的水,在該溫度下繼續脫水縮合反應1小時,然后將反應物冷卻。如上所述,合成重均分子量為18,000、酸值為15mgkoh/g且玻璃化轉變溫度為60℃的聚酯樹脂(1)。基于jisk0070-1992通過中和滴定法測量樹脂的酸值。
將40份乙酸乙酯和25份2-丁醇放入設置由溫度調節器和氮置換單元的容器中以獲得混合溶劑,隨后向其中緩慢倒入100份聚酯樹脂(1)并使其在其中溶解,向其放入10%氨水溶液(以相對于樹脂酸值的摩爾比為3倍當量),并將混合物攪拌30分鐘。
然后,將容器的內含物用干燥氮氣置換,將溫度保持在40℃,在將混合溶液攪拌的同時以2份/分鐘的速度滴加400份離子交換水,由此進行乳化。在完成滴加之后,使乳化溶液的溫度返回室溫(20℃~25℃),通過在攪拌乳化溶液的同時用干燥氮氣進行48小時鼓泡而將乙酸乙酯和2-丁醇降低至1,000ppm以下,由此獲得分散有體積平均粒徑為200nm的樹脂顆粒的樹脂顆粒水分散液。對樹脂顆粒水分散液添加離子交換水,將固體含量調節至20%,由此獲得樹脂顆粒水分散液(1)。
光輝性顏料水分散液(1)的制備
·光輝性顏料分散液(1):100份
·陰離子型表面活性劑(由dskco.,ltd.制造的neogenr):1.5份
·離子交換水:900份
利用乳化分散器cavitron(由pacificmachinery&engineeringco.,ltd.制造的cr1010)將上述材料混合分散1小時,通過在攪拌所述材料的同時用干燥氮氣進行48小時鼓泡而將ipa降低至1,000ppm以下,由此獲得其中分散有光輝性顏料(鋁顏料)的光輝性顏料水分散液(1)(固體含量:10%)。
防粘劑水分散液(1)的制備
·石蠟(由nipponseiroco.,ltd.制備的hnp-9):100份
·陰離子型表面活性劑(由dskco.,ltd.制造的neogenrk):1份
·離子交換水:350份
將上述成分混合并在100℃加熱,使用均質器(由ika制造的ultra-turraxt50)分散,并利用mantongaulin高壓均質器(由mantongaulinmanufacturingco.,inc.制造)進行分散處理,由此獲得其中分散有體積平均粒徑為200nm的防粘劑顆粒的防粘劑水分散液(1)(固體含量:20%)。
色調劑的制備
·樹脂顆粒水分散液(1):450份
·防粘劑水分散液(1):50份
·光輝性顏料水分散液(1):21.7份
·非離子型表面活性劑(igepalca897):1.4份
將上述原料置于2l圓筒形不銹鋼容器中,在通過均質器(由ika制造的ultra-turraxt50)以4000rpm施加剪切力的同時分散并混合10分鐘。然后,向其中緩慢滴加聚氯化鋁的10%硝酸水溶液1.75份作為凝集劑,通過將均質器的旋轉速度設定在5000rpm而將該混合物分散并混合15分鐘,由此獲得原料分散液。
其后,將原料分散液轉移到設置有利用雙槳片攪拌槳葉的攪拌器和溫度計的聚合槽中,在將攪拌旋轉速度設定為550rpm的同時開始通過覆套式加熱器加熱,并在54℃促進凝集顆粒的生長。此時,通過0.3n的硝酸和1n的氫氧化鈉水溶液將原料分散液的ph控制在2.2~3.5的范圍內。原料分散液在該ph范圍內保持約2小時,形成凝集顆粒。此時,利用multisizerii(孔徑:50μm,由beckmancoulter,inc.制造)測量的凝集顆粒的體積平均粒徑為10.6μm。
接下來,額外添加100份樹脂顆粒水分散液(1),使樹脂顆粒附著于凝集顆粒的表面。將溫度進一步升高至56℃,并在通過光學顯微鏡和multisizerii觀察粒徑和構造的同時使凝集顆粒組織化。
其后,將ph增加至8.0以使凝集顆粒聚并,然后以0.01℃/分鐘的速度將溫度升高至80℃。在通過光學顯微鏡檢查出凝集顆粒已聚并之后,在將溫度維持在80℃的同時將ph降至6.0,在2.5小時后停止加熱,并以1.0℃/分鐘的降溫速度進行冷卻。其后,將顆粒用20μm的篩網分級,用水反復洗滌,并通過真空干燥器干燥,由此獲得光輝性色調劑顆粒(1)。
表1
測量/評估
甲苯不溶性部分的測量
通過所述方法測量各實施例獲得的光輝性色調劑中的甲苯不溶性部分(除光輝性顏料和外添劑之外的甲苯不溶性部分)。結果將在表2中示出。
顯影劑的制備
將36份各實施例中獲得的光輝性色調劑和414份載體放入2l共混器中并攪拌20分鐘,隨后將212μm的混合物分級以制備各顯影劑。作為載體,使用通過下述方法獲得的載體。
載體的制備
·鐵氧體顆粒(體積平均粒徑:35μm):100份
·甲苯:14份
·甲基丙烯酸甲酯-丙烯酸全氟辛基乙酯共聚物(臨界表面張力:24dyn/cm):1.6份
·炭黑(產品名稱:由cabotcorporation制造的vxc-72,體積電阻率:等于或小于100ωcm):0.12份
·交聯的三聚氰胺樹脂顆粒(平均粒徑:0.3μm,不溶于甲苯):0.3份
首先,將炭黑在甲苯中稀釋,將混合物添加至甲基丙烯酸甲酯-丙烯酸全氟辛基乙酯共聚物并在砂磨機中分散。隨后,通過攪拌器將除鐵氧體顆粒以外的上述各成分分散10分鐘,獲得覆蓋層形成用溶液。隨后,將覆蓋層形成用溶液和鐵氧體顆粒放入真空脫氣型捏合機中,并將混合物在60℃攪拌30分鐘。然后,降低壓力,將甲苯蒸發,形成樹脂覆蓋層,由此獲得載體。
顆粒度
用所得顯影劑填充由fujixeroxco.,ltd.制造的“改進顏色800press”的顯影機。在填充顯影劑之后,將顯影機在80%rh的環境中于35℃保持72小時。
使用該改進機器來在80%rh的環境中于35℃下在10,000張coat紙(基重:127,由ojipaperco.,ltd.制造)上打印色調劑圖像,其包含光輝性色調劑施加量為3.5g/m2的實心圖像部分和光輝性色調劑導致的灰度圖像部分。視覺觀察第10,000個色調劑圖像的灰度圖像部分中的顆粒度,并基于以下標準評估。所得結果將在表2中示出。
標準
g1:沒有識別到顆粒度
g2:略微識別到顆粒度
g3:雖然識別到顆粒度,但沒有觀察到問題
g4:識別到顆粒度,且獲得違和感
g5:強烈識別到顆粒度
比例(x/y)
使用由nippondenshokuindustriesco.,ltd.制造的分光式變角色差儀gc5000l作為可變角光度計,從而以相對第10,000個打印色調劑圖像的實心圖像部分為-45°的入射角使入射光入射在該實心圖像上,并且測量受光角+30°處的反射率x和受光角-30°處的反射率y以20nm的間隔由400nm~700nm的波長范圍內的光測量反射率x和反射率y,并且獲得各波長處的反射率的平均值。由這些測量結果計算比例(x/y)。結果將在表2中示出。
隨著比例(x/y)增加,光澤感增加。隨著比例(x/y)降低,暗色調增加,且光澤感降低。
表2也示出了色調劑顆粒的體積平均粒徑和長徑比。
表2
提供對本發明示例性實施方式的前述描述是為了說明和描述的目的。并非試圖窮盡本發明或將本發明限制于所披露的精確形式。顯然,許多改進和變化對于本領域技術人員將是顯而易見的。選擇并描述所述實施方式是為了能夠最好地解釋本發明的原理及其實際用途,由此使得本領域的其他技術人員能夠理解適用于預計的特定用途的本發明的各種實施方式和各種改進方案。試圖使本發明的范圍由下述權利要求及其等同物所限定。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 基于景深的圖像飽和度的處理方法、處理裝置和電子裝置與流程
- 一種對抗色調映射的高動態范圍圖像水印方法與流程
- 色調劑、靜電荷圖像顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法與流程
- 光輝性色調劑、靜電荷圖像顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法與流程
- 色調劑、靜電荷圖像顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法與流程
- 色調劑、顯影劑、色調劑盒、處理盒、圖像形成設備和圖像形成方法與流程
- 一種基于平均飽和度先驗的圖像去霧方法與流程
- 色調劑、顯影劑、色調劑盒、顯影劑盒、處理盒、圖像形成裝置和方法與流程
- 對圖像進行逆色調映射的方法和設備與流程
- 色調劑、顯影劑、色調劑盒、顯影劑盒、處理盒、圖像形成裝置和圖像形成方法與流程