一種聚合物/相變微膠囊復合纖維及其制備方法與流程
本發明屬于復合纖維領域,涉及一種聚合物/相變微膠囊復合纖維及其制備方法。
背景技術:
相變微膠囊是利用膠囊技術將相變材料,如石蠟,包裹在微膠囊壁材,如聚甲基丙烯酸,內部制成的相變復合材料,相變微膠囊直徑一般在微米級別,其常可做為添加劑加入到基體材料中制成相變功能性復合材料,相變功能性復合材料被廣泛應用于服裝和醫療用品等領域。
利用相變微膠囊制備相變纖維,相變纖維在30℃左右具有高溫吸熱,低溫放熱的現象,用作織物具有實時調溫的作用。
已有的相變纖維有腈綸,纖維素纖維體系,因熔紡溫度高,易導致微膠囊破損,因此溶液紡是制備相變纖維的主要方法,而纖維素的資源豐富,易于自然降解等特性也使其功能化具有更高的價值。
研究人員常在粘膠纖維或其它復合纖維中添加相變微膠囊,添加量為20%~40%時,相變焓可達10~20j/g,可以極大的提高復合纖維的品質。但在制備相變微膠囊復合纖維的過程中,往往將相變微膠囊直接與紡絲液混合,此時,由于紡絲液粘度高,使相變微膠囊難以在纖維素溶液中均勻分散,對混合設備的要求也較為苛刻,在制備過程中容易產生相變微膠囊分散不均勻和易破損流失等問題,例如文獻dissolutionandformingofcellulosewithionicliquids中纖維素聚合度為569,濃度為12.9wt%的纖維素/離子液體溶液,溫度為85℃時的零切粘度為8670pa·s;纖維素聚合度為569,濃度為10.4wt%的纖維素/1-丁基-3-甲基咪唑氯鹽([bmim]cl)溶液,溫度為85℃時的零切粘度達到7920pa·s。
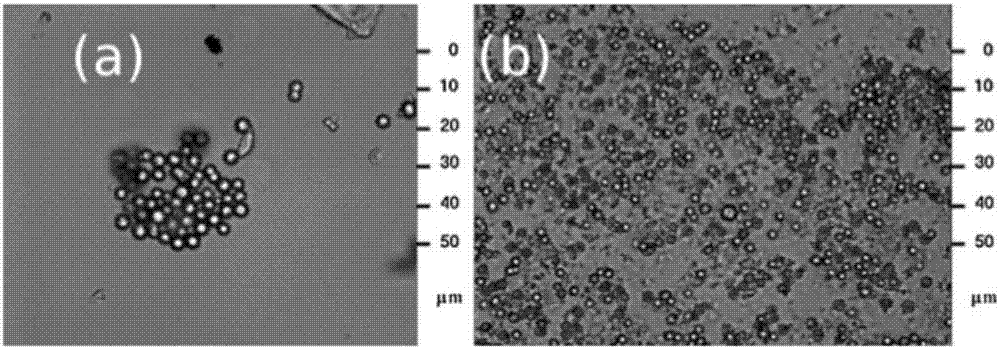
對于這一問題,也曾有其他學者指出,例如文獻fabricationandmorphologicalcharacterizationofmicroencapsulatedphasechangematerials(micropcms)andmacrocapsulescontainingmicropcmsforthermalenergystorage曾指出在相變微膠囊的制備過程中容易產生破損的情況,如圖1所示,其在制備過程中便存在一定的破損率,破損后的微膠囊中相變物質流失,失去相變功能,且小分子相變物質進入纖維中易使纖維的力學性能變差,因此,在后續制備纖維的過程中高溫和真空攪拌更易使微膠囊出現破損的情況,最終導致微膠囊的添加效率變低;文獻thermoregulatingresponseofcottonfabriccontainningmicroencapsulatedphasechangematerials中通過微膠囊的技術制備了纖維素纖維相變材料,添加過程中采用高速攪拌使微膠囊在高粘溶液中分散,如圖2所示,攪拌速率達到10000rpm時尚不能使微膠囊良好的分散,攪拌速率達13500rpm時微膠囊才可以達到分散狀態,在高攪拌速率保證其分散性良好的條件下,相變微膠囊本身的完整性則不能夠得到保證。
以上相變纖維的制備過程中存在的問題為:
(1)相變粘膠纖維強度較低;
(2)離子液體體系沒有實現工業化,且溶劑本身粘度大,給微膠囊的分散造成困難;
(3)常規分散微膠囊通常需要用分散劑,影響氮甲基嗎啉氮氧化物的回收利用;
(4)強剪切分散和高真空度作用不會影響到溶劑的回收利用,但容易使微膠囊破裂而降低添加效率。
因此,研究一種分散性好且不易發生破損流失的聚合物/相變微膠囊復合纖維及其制備方法具有十分重要的意義。
技術實現要素:
本發明先將相變微膠囊在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中分散均勻,然后再采用低真空脫水的方式,克服了傳統制備方法中直接在高真空狀態下將相變微膠囊直接置于高粘度紡絲溶液中而導致其分散性差和易破損的缺點,提供了一種分散性好且不易發生破損流失的聚合物/相變微膠囊復合纖維及其制備方法,從而提高了相變微膠囊的添加效率和最終得到的復合纖維的力學性能。
為了達到上述目的,本發明采用的技術方案為:
一種聚合物/相變微膠囊復合纖維的制備方法,步驟如下:
(1)將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中得到分散液,現有技術中常將相變微膠囊直接置于高粘度溶液中,由于溶液粘度較大,故而相變微膠囊在其中的分散性較差;
(2)加入聚合物攪拌混合后進行脫水處理得到混合分散液,所述聚合物是指能夠溶解在濃度為87wt%的氮甲基嗎啉氮氧化物水溶液中且可進行紡絲的物質,所述脫水處理是在低真空狀態下進行的,真空壓力為-0.05~0.08mpa,此時溶液粘度為最終溶液粘度的70%;現有技術采用的真空壓力為-0.1mpa,開始抽真空過程中,微膠囊并未被固定在高粘的聚合物溶液中,在高真空度下易被攪拌和真空共同作用而破損;
(3)在高真空狀態下攪拌脫水,制得功能性紡絲液,所述高真空狀態的真空壓力的絕對值大于等于0.095mpa;
(4)紡絲制得聚合物/相變微膠囊復合纖維。
本發明先將微膠囊與溶劑氮甲基嗎啉氮氧化物混合,在低剪切條件下制備微膠囊/氮甲基嗎啉氮氧化物分散液,再在低真空狀態下進行溶解纖維素,待溶液達到一定的粘度,再用高真空脫水使纖維素完全溶解,此時溶液粘度高微膠囊在溶液中難以運動,因此高真空狀態不會破壞微膠囊的結構。
作為優選的技術方案:
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,所述聚合物為纖維素、醋酸纖維素或聚芳砜酰胺。本發明中的聚合物的材料包括但不限于此,能夠滿足溶解在濃度為87wt%的氮甲基嗎啉氮氧化物水溶液中且可進行紡絲的物質皆可作為本發明中聚合物的備選材料。
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,步驟(1)中,所述相變微膠囊的直徑為10μm,相變微膠囊中壁材與芯材的質量比為1:1,壁材為苯乙烯與甲基丙烯酸的共聚物,芯材為質量比為1:1的正十八烷和硬脂酸丁酯的混合物,所述分散液中相變微膠囊相對于濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的含量為0.2~1wt%。
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,步驟(1)中,將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中采用的方式為在線添加,即在生產線上的濃度為50wt%的氮甲基嗎啉氮氧化物水溶液輸送管路上加裝微膠囊在線添加裝置。
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,步驟(2)中,所述脫水處理采用的方式為減壓蒸餾;所述脫水處理的溫度為80~110℃;所述混合分散液中聚合物的含量為2~6wt%,聚合物與相變微膠囊的質量比為5~20:1。
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,步驟(3)中,所述攪拌脫水采用的方式為減壓蒸餾,所述攪拌脫水的溫度為80~110℃,所述功能性紡絲液中聚合物的含量為6~12wt%,所述高真空狀態的真空壓力為-0.095~0.1mpa。
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,步驟(4)中,所述紡絲采用的工藝為濕法紡絲工藝或干噴濕法紡絲工藝。
如上所述的一種聚合物/相變微膠囊復合纖維的制備方法,所述紡絲的工藝流程為擠出、凝固和牽伸,其中凝固是在濃度為0~10wt%nmmo水溶液中進行的,紡絲工藝參數為:卷繞速率5~50m/min,牽伸倍數5~10。
本發明還提供了與上述制備方法相對應的一種聚合物/相變微膠囊復合纖維,相變微膠囊在纖維中分散均勻,纖維可以是長絲,也可以是短纖維,具體表現為復合纖維中相變微膠囊的平均粒徑為10~15μm,復合纖維的干態斷裂強度為1.7~4.5cn/dtex;相變微膠囊在加工過程中不容易發生破損流失,具體表現為相變微膠囊的添加效率為90~95%,所述添加效率的具體計算公式為:
常規方法一般為60%左右。
作為優選的技術方案:
如上所述的聚合物/相變微膠囊復合纖維,所述聚合物/相變微膠囊復合纖維的單絲纖度為0.8~4.8dtex。
發明原理:
本發明先將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中,濃度為50wt%的氮甲基嗎啉氮氧化物水溶液更易與相變微膠囊均勻混合,保證相變微膠囊分散均勻后,再加入聚合物漿粕攪拌混合并進行低真空脫水,此時體系還未形成高粘溶液,微膠囊在混合分散液中仍處于自由分散狀態,低真空度既能促進相變微膠囊與聚合物溶液的均勻混合又能有效防止將微膠囊直接置于高溫真空狀態下混合而導致其破損的現象,待溶液粘度逐漸升高,再進行高真空度脫水,此時微膠囊被固定包埋在高粘溶液當中,微膠囊不易破損。本發明克服了傳統制備方法中直接在高真空狀態下將相變微膠囊直接置于高粘度紡絲溶液中導致其分散性差和易破損的缺點,在保證相變微膠囊分散均勻的同時減少了相變微膠囊的破損流失現象,從而同時提高了相變微膠囊的添加效率和最終形成的纖維的力學性能。
有益效果:
(1)本發明的一種聚合物/相變微膠囊復合纖維的制備方法,僅需在聚合物/相變微膠囊復合纖維生產線上增加在線添加裝置即可制得分散性良好的聚合物/相變微膠囊復合纖維,無需額外分散劑的加入,不增加溶劑與凝固浴回收的負擔,成本低,方法簡便靈活。
(2)本發明的一種聚合物/相變微膠囊復合纖維的制備方法,相變微膠囊同時具有了分散性良好和不易破損的優點,采用低粘度溶液與低真空脫水再配合高真空脫水的方式,克服了傳統制備方法中高粘度溶液相變微膠囊分散性差和高真空狀態下相變微膠囊易破損的缺點。
(3)本發明的一種聚合物/相變微膠囊復合纖維,復合纖維中相變微膠囊分散性好且破損率低,從而進一步提高了相變微膠囊的添加效率和最終得到的復合纖維的力學性能。
附圖說明
圖1為電鏡下不同放大倍數下相變微膠囊的破損情況示意圖,其中圖a和圖b為以苯乙烯-二乙烯基苯共聚物為壁材的微膠囊,圖a的放大倍數為4000,圖b的放大倍數為16000;圖c和圖d為以聚胺酯為壁材的微膠囊,圖c的放大倍數為30000,圖d的放大倍數為45000;
圖2為相變微膠囊在高粘溶液中的分散狀態示意圖,其中圖a為轉速為10000rpm的分散狀態示意圖,圖b為轉速13500rpm的分散狀態示意圖。
具體實施方式
下面結合具體實施方式,進一步闡述本發明。應理解,這些實施例僅用于說明本發明而不用于限制本發明的范圍。此外應理解,在閱讀了本發明講授的內容之后,本領域技術人員可以對本發明作各種改動或修改,這些等價形式同樣落于本申請所附權利要求書所限定的范圍。
實施例1
一種纖維素/相變微膠囊復合纖維的制備方法,步驟如下:
(1)在纖維素/相變微膠囊復合纖維的濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的輸送管路上加裝微膠囊在線添加裝置,通過在線添加的方式將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中得到分散液,相變微膠囊的直徑為10μm,相變微膠囊中壁材與芯材的質量比為1:1,壁材為苯乙烯與甲基丙烯酸的共聚物,芯材為質量比為1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相變微膠囊相對于濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的含量為0.8wt%;
(2)加入纖維素攪拌混合后在真空壓力為-0.07mpa的條件下通過減壓蒸餾的方式進行脫水處理得到混合分散液,脫水處理的溫度為80℃,混合分散液中纖維素的含量為6wt%,纖維素與相變微膠囊的質量比為7:1;
(3)在真空壓力為-0.095mpa的條件下通過減壓蒸餾的方式進行攪拌脫水制得功能性紡絲液,攪拌脫水的溫度為80℃,功能性紡絲溶液中纖維素的含量為8wt%;
(4)經濕法紡絲工藝的擠出、凝固和牽伸后制得纖維素/相變微膠囊復合纖維,其中凝固在水中進行,卷繞速率為10m/min,牽伸倍數為7。
最終制得的纖維素/相變微膠囊復合纖維的干態斷裂強度為3cn/dtex,單絲纖度為3dtex,其中相變微膠囊的平均粒徑為12μm,添加效率為92%。
實施例2
一種纖維素/相變微膠囊復合纖維的制備方法,步驟如下:
(1)在纖維素/相變微膠囊復合纖維的濃度為50wt%的氮甲基嗎啉氮氧化物氮甲基嗎啉氮氧化物水溶液的輸送管路上加裝微膠囊在線添加裝置,通過在線添加的方式將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中得到分散液,相變微膠囊的直徑為10μm,相變微膠囊中壁材與芯材的質量比為1:1,壁材為苯乙烯與甲基丙烯酸的共聚物,芯材為質量比為1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相變微膠囊相對于濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的含量為1wt%;
(2)加入纖維素攪拌混合后在真空壓力為-0.05mpa的條件下通過減壓蒸餾的方式進行脫水處理得到混合分散液,脫水處理的溫度為110℃,混合分散液中纖維素的含量為3wt%,纖維素與相變微膠囊的質量比為5:1;
(3)在真空壓力為-0.1mpa的條件下通過減壓蒸餾的方式進行攪拌脫水制得功能性紡絲液,攪拌脫水的溫度為110℃,功能性紡絲溶液中纖維素的含量為6wt%;
(4)經濕法紡絲工藝的擠出、凝固和牽伸后制得纖維素/相變微膠囊復合纖維,其中凝固在濃度為10wt%的氮甲基嗎啉氮氧化物水溶液中進行,卷繞速率為5m/min,牽伸倍數為10。
最終制得的纖維素/相變微膠囊復合纖維的干態斷裂強度為4.5cn/dtex,單絲纖度為4.8dtex,其中相變微膠囊的平均粒徑為15μm,添加效率為93%。
對比例1
將與實施例2相同的相變微膠囊分散在與實施2相同的纖維素的氮甲基嗎啉氮氧化物水溶液攪拌混合均勻得到分散液,分散液中相變微膠囊、纖維素、氮甲基嗎啉氮氧化物和水的比例關系同實施例1中步驟(1)制得的分散液,然后將分散液在真空壓力為-0.1mpa的條件下通過減壓蒸餾的方式進行攪拌脫水制得功能性紡絲液,攪拌脫水的溫度為110℃,功能性紡絲溶液中纖維素的含量為6wt%,然后采用與實施例1步驟(4)相同的紡絲工藝和紡絲參數紡絲制得纖維素/相變微膠囊復合纖維。
最終制得的纖維素/相變微膠囊復合纖維的干態斷裂強度為1.6cn/dtex,單絲纖度為0.8dtex,其中相變微膠囊的平均粒徑為5μm,添加效率為62%。將實施例2與對比例1相對比可以看出,本發明先將相變微膠囊在濃度為50wt%的氮甲基嗎啉氮氧化物中分散均勻,然后再采用低真空脫水的方式,克服了對比例1中直接在高真空狀態下將相變微膠囊直接置于高粘度紡絲溶液中而導致其分散性差和易破損的缺點,極大的改善了相變微膠囊的分散均勻性,同時,待溶液粘度升高后,微膠囊被固定包埋在高粘溶液當中,微膠囊破損率低,因此,使得最終制得的復合纖維的相變微膠囊的添加效率和纖維本身的力學性能相對于比對比例1有了明顯提高。
實施例3
一種聚芳砜酰胺/相變微膠囊復合纖維的制備方法,步驟如下:
(1)在聚芳砜酰胺/相變微膠囊復合纖維的濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的輸送管路上加裝微膠囊在線添加裝置,通過在線添加的方式將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中得到分散液,相變微膠囊的直徑為10μm,相變微膠囊中壁材與芯材的質量比為1:1,壁材為苯乙烯與甲基丙烯酸的共聚物,芯材為質量比為1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相變微膠囊相對于濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的含量為0.2wt%;
(2)加入聚芳砜酰胺攪拌混合后在真空壓力為-0.08mpa的條件下通過減壓蒸餾的方式進行脫水處理得到混合分散液,脫水處理的溫度為90℃,混合分散液中聚芳砜的含量為6wt%,聚芳砜與相變微膠囊的質量比為20:1;
(3)在真空壓力為-0.1mpa的條件下通過減壓蒸餾的方式進行攪拌脫水制得功能性紡絲液,攪拌脫水的溫度為90℃,功能性紡絲溶液中聚芳砜酰胺的含量為15wt%;
(4)經干噴濕法紡絲工藝的擠出、凝固和牽伸后制得聚芳砜酰胺/相變微膠囊復合纖維,其中凝固在濃度為5wt%的氮甲基嗎啉氮氧化物水溶液中進行,卷繞速率為50m/min,牽伸倍數為5。
最終制得的聚芳砜酰胺/相變微膠囊復合纖維的干態斷裂強度為1.7cn/dtex,單絲纖度為0.8dtex,其中相變微膠囊的平均粒徑為10μm,添加效率為90%。
實施例4
一種醋酸纖維素/相變微膠囊復合纖維的制備方法,步驟如下:
(1)在醋酸纖維素/相變微膠囊復合纖維的濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的輸送管路上加裝微膠囊在線添加裝置,通過在線添加的方式將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中得到分散液,相變微膠囊的直徑為10μm,相變微膠囊中壁材與芯材的質量比為1:1,壁材為苯乙烯與甲基丙烯酸的共聚物,芯材為質量比為1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相變微膠囊相對于濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的含量為0.6wt%;
(2)加入醋酸纖維素攪拌混合后在真空壓力為-0.065mpa的條件下通過減壓蒸餾的方式進行脫水處理得到混合分散液,脫水處理的溫度為98℃,混合分散液中醋酸纖維素的含量為7wt%,醋酸纖維素與相變微膠囊的質量比為7.5:1;
(3)在真空壓力為-0.096mpa的條件下通過減壓蒸餾的方式進行攪拌脫水制得功能性紡絲液,攪拌脫水的溫度為98℃,功能性紡絲溶液中醋酸纖維素的含量為9wt%;
(4)經干噴濕法紡絲工藝的擠出、凝固和牽伸后制得醋酸纖維素/相變微膠囊復合纖維,其中凝固在濃度為8wt%的氮甲基嗎啉氮氧化物水溶液中進行,卷繞速率為15m/min,牽伸倍數為10。
最終制得的醋酸纖維素/相變微膠囊復合纖維的干態斷裂強度為3.0cn/dtex,單絲纖度為2.8dtex,其中相變微膠囊的平均粒徑為13.5μm,添加效率為94%。
實施例5
一種纖維素/相變微膠囊復合纖維的制備方法,步驟如下:
(1)在纖維素/相變微膠囊復合纖維的濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的輸送管路上加裝微膠囊在線添加裝置,通過在線添加的方式將相變微膠囊分散在濃度為50wt%的氮甲基嗎啉氮氧化物水溶液中得到分散液,相變微膠囊的直徑為10μm,相變微膠囊中壁材與芯材的質量比為1:1,壁材為苯乙烯與甲基丙烯酸的共聚物,芯材為質量比為1:1的正十八烷和硬脂酸丁酯的混合物,分散液中相變微膠囊相對于濃度為50wt%的氮甲基嗎啉氮氧化物水溶液的含量為0.4wt%;
(2)加入纖維素攪拌混合后在真空壓力為-0.07mpa的條件下通過減壓蒸餾的方式進行脫水處理得到混合分散液,脫水處理的溫度為105℃,混合分散液中纖維素的含量為10wt%,纖維素與相變微膠囊的質量比為13:1;
(3)在真空壓力為-0.098mpa的條件下通過減壓蒸餾的方式進行攪拌脫水制得功能性紡絲液,攪拌脫水的溫度為105℃,功能性紡絲溶液中纖維素的含量為12.5wt%;
(4)經干噴濕法紡絲工藝的擠出、凝固和牽伸后制得纖維素/相變微膠囊復合纖維,其中凝固在濃度為9wt%的氮甲基嗎啉氮氧化物水溶液中進行,卷繞速率為20m/min,牽伸倍數為8。
最終制得的纖維素/相變微膠囊復合纖維的干態斷裂強度為3.2cn/dtex,單絲纖度為3.0dtex,其中相變微膠囊的平均粒徑為11μm,添加效率為90%。
- 還沒有人留言評論。精彩留言會獲得點贊!