一種SnO2/ZnO復合微納米纖維的制備方法及其產品與流程
本發明涉及一種sno2/zno復合微納米纖維的制備方法,具體涉及一種纖維表面具有sno2顆粒的尺寸可調的sno2/zno復合微納米纖維的制備方法。
背景技術:
基于半導體氧化物的氣敏傳感器能夠快速檢測出易制毒和易燃氣體,近年來受到了環境治理和工業安全領域越來越多的關注。合成靈敏度高、穩定性強、檢測限低的氣敏傳感器是現階段該領域研究的重點問題。研究表明,具有不同微觀形貌特征的半導體氣敏傳感器常展現出獨特的物理化學性質。因此,利用合適的制備工藝得到形貌與尺寸可控,成分可調的微納米半導體氣敏傳感器具有重要的應用價值。
sno2與zno都是典型的n型半導體,直接禁帶寬度分別為3.6和3.37ev,具有高溫化學穩定性好、氣體靈敏度高、特殊的光學、催化和電學性能的特點,廣泛應用于傳統的氣體傳感領域中。國內外的研究發現,通過選擇適宜的合成方法,如水熱法、微波輔助法、靜電紡絲法等能夠有效制備具有不同形貌特點的sno2與zno微納米結構(如類球形顆粒、納米棒、中空微球、立方塊、一維纖維等)。通過摻雜或異質結構筑等方式得到的sno2/zno微納米材料,不僅能夠形成有效的n-n異質結,還顯示出兩種氧化物的氣敏協同作用,通常具有更加優異的氣敏特性。例如,“q.ge,s.y.ma,y.b.xu,x.l.xu,h.chen,z.qiang,h.m.yang,andq.z.zeng,mater.lett.,2017,191,5-9”報道了利用兩步合成法制備了zno/sno2納米花結構,發現該復合納米材料對甲醇具有良好的氣敏檢測性能。
利用靜電紡絲法制備一維半導體氧化物微納米材料具有許多優點,如紡絲參數易于調控,制備的一維材料比表面積大,直徑可調,載流子的輸運過程沿一維線性方向速度快,氣體靈敏性顯著等,得到了國內外學者的青睞。利用常規靜電紡絲法制備sno2/zno復合微納米纖維的過程主要包括:(1)直接將錫鹽和鋅鹽混合均勻形成前驅體紡絲液,調節靜電紡絲過程得到前驅體纖維,再經過熱處理過程得到最終復合纖維結構。該方法得到的復合纖維整體由sno2顆粒和zno顆粒均勻分布組成,常伴有團聚現象,主要由熱處理制度控制sno2顆粒和zno顆粒的尺寸范圍;(2)首先利用靜電紡絲和熱處理過程得到sno2或zno微納米纖維,然后通過水熱合成、超聲、高溫回流法等方式在纖維表面上負載第二相納米材料。該方法得到的纖維制備工藝相對復雜,形貌重復性差,纖維的直徑只受靜電紡絲和熱處理過程影響,表面異質相的尺寸和分布狀況只受第二步合成參數影響。利用靜電紡絲過程制備的具有表面異質結的sno2/zno微納米纖維大都使用兩步合成法,即通過水熱合成、超聲、高溫回流法等方式在靜電紡絲法得到的纖維表面上負載第二相納米材料。上述合成過程較為復雜,且一維連續纖維容易斷裂,表面異質結的穩定性差。
技術實現要素:
本發明的目的是提供一種合成簡單,原料成本低廉,產物形貌與尺寸可控的sno2/zno復合微納米纖維的制備方法以及使用該方法制造的表面具有sno2顆粒的sno2/zno復合微納米纖維。
為實現上述目的,本發明采用如下技術方案。
一種sno2/zno復合微納米纖維的制備方法,包括以下步驟:
(1)將一定量的鋅鹽、2-甲基咪唑分別溶于甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌一段時間,經過靜置、離心洗滌、干燥后得到zif-8產物;
(2)將一定量的檸檬酸氫二銨、十八胺、乙基纖維素、zif-8溶于乙醇溶劑中,攪拌得到透明溶液;
(3)將一定量的錫鹽和聚乙烯吡咯烷酮(pvp)溶于乙醇和二甲基甲酰胺(dmf)的混合溶劑中,攪拌得到透明溶液,然后緩慢滴加到步驟(2)得到的溶液中,攪拌得到前驅體紡絲液;
(4)將前驅體紡絲液通過靜電紡絲形成前驅體纖維,然后將前驅體纖維進行熱處理,得到sno2/zno復合微納米纖維。
上述步驟(1)中,鋅鹽和2-甲基咪唑的摩爾比為1:3.0-4.5,鋅鹽在甲醇溶液中的濃度為0.045-0.055mol/l;
上述步驟(1)中,所述鋅鹽為鋅的硝酸鹽;鋅鹽和2-甲基咪唑的甲醇溶液混合均勻后攪拌1-2h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干。
上述步驟(2)中,zif-8、檸檬酸氫二銨、十八胺和乙基纖維素單體的摩爾比為1:0.50-0.80:0.80-1.25:2.0-3.0,zif-8在乙醇溶劑中的濃度為0.002-0.120mol/l。
上述步驟(3)中,所述錫鹽為錫的鹵化物;錫鹽和pvp的摩爾比為1:5.5-7.0,乙醇和dmf的體積比為1:4,錫鹽在乙醇和dmf的混合溶劑中的濃度為0.16-0.24mol/l,其中聚乙烯吡咯烷酮的摩爾量按其聚合單體的摩爾量計;
上述步驟(3)中,所述zif-8溶液與錫鹽溶液的體積比為1:2.2-3.2,兩種溶液混合時的滴加速度為0.2-0.3ml/min,兩種溶液完全混合后,繼續室溫攪拌8-10h。
上述步驟(4)中,靜電紡絲時,正電壓為16-20kv,負電壓為0.5kv,接收距離為18-22cm,注射器推進速度為0.001-0.003mm/s;
上述步驟(4)中,熱處理過程是:將前驅體纖維按照1-2℃/min的升溫速度由室溫升至550-650℃,保溫1-4h。
一種上述方法生產的sno2/zno復合微納米纖維,主體纖維是由sno2和zno納米顆粒復合而成,表面具有sno2納米顆粒;纖維直徑為0.05-1.5μm;表面wo3顆粒呈球狀,球狀結構的直徑為0.05-2.5nm。
本發明首先將一定量的鋅鹽和2-甲基咪唑的甲醇溶液混合均勻,室溫下合成zif-8納米顆粒,然后將一定量的zif-8顆粒與檸檬酸氫二銨、十八胺和乙基纖維素同時溶于乙醇溶劑中,實現對zif-8顆粒表面的有效改性,提高zif-8顆粒的穩定性,并且選擇加入的檸檬酸氫二銨、十八胺和乙基纖維素這三種功能添加劑的種類和加入比例直接影響復合纖維的最終形貌,即步驟(2)中功能添加劑的使用對產物的無機相成核長大過程及其最終形貌具有重要作用。通過與步驟(3)中的溶液混合均勻,嚴格控制混合滴加速度,能夠保持混合溶液的物化性能穩步改善,最終得到zif-8與sn鹽共存溶液體系,利用合適比例的pvp、乙醇和dmf混合均勻后具有的成纖性能,調控紡絲參數得到復合前驅體纖維,經過適當的熱處理過程最終得到纖維表面具有sno2顆粒的尺寸可調的sno2/zno復合微納米纖維。其中,pvp為導電聚合物,zif-8為鋅源和結構誘導劑。
本發明通過在前驅體紡絲液中引入一定量的表面改性zif-8顆粒及幾類重要的功能添加劑,使得到的前驅體纖維中均勻分散了zif-8納米顆粒,并且改善了前驅體纖維的成分及高溫分解與無機物成核長大過程,這種前驅體纖維成分和微觀結構的改變在特定的熱處理過程中導致了sno2/zno復合微納米纖維的成分與形貌可控性分布。隨著zif-8納米顆粒加入量的不斷增加,本發明得到的sno2/zno復合材料的主體纖維直徑逐步減小,且保持sno2/zno復合相的成分特征,而表面異質相的成分保持單一的sno2相,但sno2相顆粒逐漸增多。這種現象說明,通過在前驅體纖維中引入一定量的zif-8納米顆粒,不僅能夠在熱處理過程中調控原料中有機基團的分解反應、氧化還原反應、無機化合物的成核與長大過程等,更重要的是能夠有效控制主體纖維中sno2/zno復合相的含量比例及sno2相顆粒的表面析出與長大過程,是一步熱處理過程形成纖維表面具有sno2顆粒的尺寸可調的sno2/zno復合微納米纖維的根本原因。研究發現,只有在特定含量、特定顆粒尺寸的zif-8納米顆粒的存在下才能得到本發明中的產物形貌與成分分布特征。也就是說,只有在本發明權利要求的范圍內,才能得到最終產物。
本發明具有以下有益優點:
本發明所提供的技術方法新穎,產物形貌特殊,反應機理與傳統sno2/zno復合材料的合成機理存在本質不同。本發明首次將表面修飾的zif-8結構引入靜電紡絲技術制備sno2/zno復合微納米纖維的領域中,反應進程可控,操作便捷,產物結構特點鮮明,形貌特殊且可控,重復性好,具有優良的氣敏性能。
附圖說明
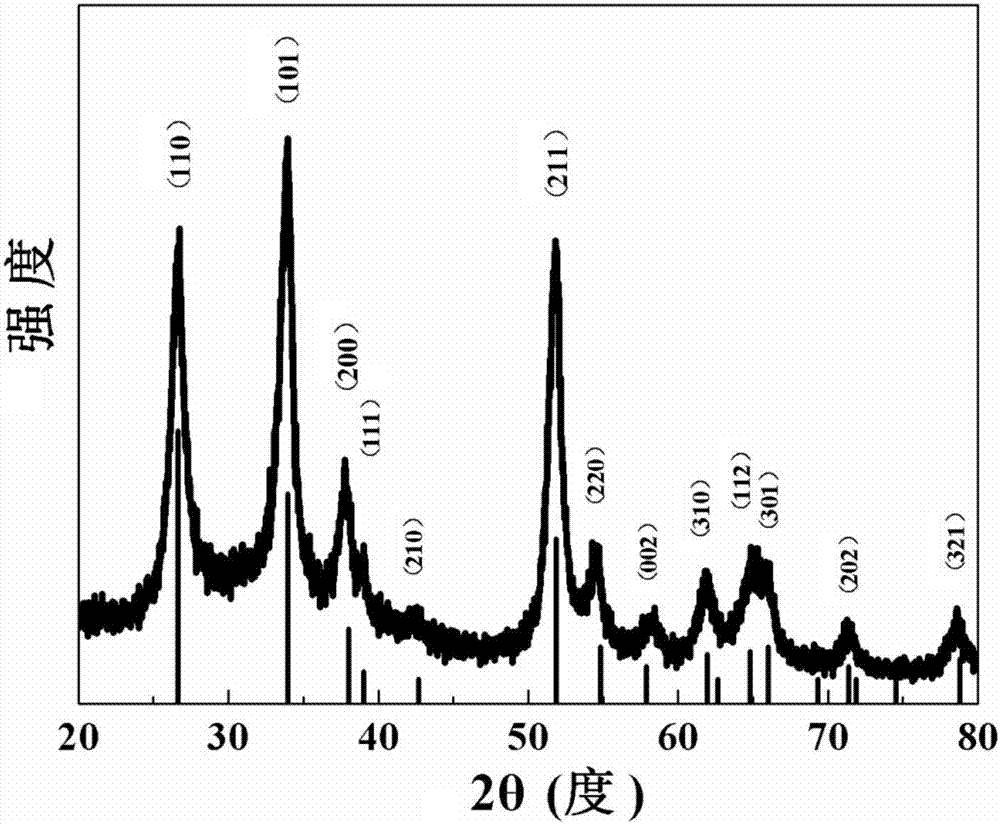
圖1為本發明實施例1合成的sno2/zno復合微納米纖維的x射線衍射(xrd)圖譜;
圖2為本發明實施例1合成的sno2/zno復合微納米纖維的掃描電鏡(sem)圖片;
圖3為本發明實施例1合成的sno2/zno復合微納米纖維的sem圖片;
圖4為本發明實施例1合成的sno2/zno復合微納米纖維的透射電鏡(tem)圖片;
圖5為本發明實施例1合成的sno2/zno復合微納米纖維的高分辨透射電鏡(hrtem)圖片。
具體實施方式
下面結合實施例和附圖對本發明做進一步說明,但本發明不受下述實施例的限制。
本發明所用pvp的分子量大于100萬,下述實施例中,所用pvp的分子量為1300000,pvp的摩爾數按單體計算,其單體摩爾質量為111。
實施例1
(1)將0.7419g的六水合硝酸鋅(zn(no3)2?6h2o)和0.8188g的2-甲基咪唑分別溶于50ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌1h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)將0.0041g的檸檬酸氫二銨、0.0082g的十八胺、0.0186g的乙基纖維素和0.0068g的zif-8、溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)將0.3541g的五水合四氯化錫(sncl4?5h2o)和0.7000g的pvp加入到1.0ml的乙醇和4.0ml的dmf的混合溶劑中,攪拌得到透明溶液,然后以0.2ml/min緩慢滴加到(2)得到的溶液中,室溫攪拌10h得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為18kv,負電壓為0.5kv,接收距離為20cm,注射器推進速度為0.002mm/s;
(4)將前驅體纖維置于馬弗爐中,按照1℃/min的升溫速度由室溫升至600℃,保溫2h,樣品隨爐冷卻后得到sno2/zno復合微納米纖維。
產物的xrd結果如圖1所示,從圖中可以看出,所有的衍射峰均與標準xrd卡(41-1445)保持一致,證明所得產物為四方相的sno2,由于zif-8的添加量較低,受xrd檢測限的影響,xrd結果中沒有發現明顯的zno晶相;產物的sem如圖2和3所示,從圖中可以看出,本發明得到的產物是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構,主體纖維是由sno2和zno納米顆粒復合而成,纖維直徑為0.12-0.14μm,纖維表面球狀結構是由sno2納米顆粒組成,球狀結構的直徑為0.12-0.17μm;產物的tem如圖4所示,從圖中可以看出,樣品形貌與sem結果基本保持一致;產物的hrtem如圖5所示,從圖中可以看出,主體纖維部分是由sno2和zno納米顆粒復合而成,纖維表面球狀結構是由sno2納米顆粒組成。
實施例2
(1)將0.7419g的zn(no3)2?6h2o和0.7269g的2-甲基咪唑分別溶于46ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌2h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)將0.0010g的檸檬酸氫二銨、0.0014g的十八胺和0.0037g的乙基纖維素和0.0014g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)將0.3541g的sncl4?5h2o和0.6200g的pvp加入到1.1ml的乙醇和4.4ml的dmf的混合溶劑中,攪拌得到透明溶液,然后以0.3ml/min緩慢滴加到(2)得到的溶液中,室溫攪拌10h得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為16kv,負電壓為0.5kv,接收距離為18cm,注射器推進速度為0.001mm/s;
(4)將前軀體纖維置于馬弗爐中,按照1℃/min的升溫速度由室溫升至550℃,保溫1h,樣品隨爐冷卻后得到纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構,主體纖維是由sno2和zno納米顆粒復合而成,纖維直徑為0.08-0.10μm,纖維表面球狀結構是由sno2納米顆粒組成,球狀結構的直徑為0.08-0.12μm。
實施例3
(1)將0.7419g的zn(no3)2?6h2o和0.9009g的2-甲基咪唑分別溶于48ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌1h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)將0.0325g的檸檬酸氫二銨、0.0594g的十八胺和0.1327g的乙基纖維素和0.0431g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)將0.3541g的sncl4?5h2o和0.7700g的pvp加入到1.2ml的乙醇和4.8ml的dmf的混合溶劑中,攪拌得到透明溶液,然后以0.2ml/min緩慢滴加到(2)得到的溶液中,室溫攪拌8h得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為17kv,負電壓為0.5kv,接收距離為22cm,注射器推進速度為0.003mm/s;
(4)將前軀體纖維置于馬弗爐中,按照2℃/min的升溫速度由室溫升至650℃,保溫4h,樣品隨爐冷卻后得到纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構,主體纖維是由sno2和zno納米顆粒復合而成,纖維直徑為0.95-1.2μm,纖維表面球狀結構是由sno2納米顆粒組成,球狀結構的直徑為1.7-2.2μm。
實施例4
(1)將0.7419g的zn(no3)2?6h2o和0.7781g的2-甲基咪唑分別溶于53ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌1h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)將0.0117g的檸檬酸氫二銨、0.0212g的十八胺和0.0506g的乙基纖維素和0.0210g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)將0.3541g的sncl4?5h2o和0.6500g的pvp加入到0.9ml的乙醇和3.6ml的dmf的混合溶劑中,攪拌得到透明溶液,然后以0.3ml/min緩慢滴加到(2)得到的溶液中,室溫攪拌8h得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為19kv,負電壓為0.5kv,接收距離為21cm,注射器推進速度為0.002mm/s;
(4)將前軀體纖維置于馬弗爐中,按照1℃/min的升溫速度由室溫升至600℃,保溫3h,樣品隨爐冷卻后得到纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構,主體纖維是由sno2和zno納米顆粒復合而成,纖維直徑為0.35-0.55μm,纖維表面球狀結構是由sno2納米顆粒組成,球狀結構的直徑為0.45-0.80μm。
實施例5
(1)將0.7419g的zn(no3)2?6h2o和0.8600g的2-甲基咪唑分別溶于51ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌2h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)將0.0083g的檸檬酸氫二銨、0.0139g的十八胺和0.0322g的乙基纖維素和0.0105g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)將0.3541g的sncl4?5h2o和0.6900g的pvp加入到1.0ml的乙醇和4.0ml的dmf的混合溶劑中,攪拌得到透明溶液,然后以0.3ml/min緩慢滴加到(2)得到的溶液中,室溫攪拌10h得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為17kv,負電壓為0.5kv,接收距離為19cm,注射器推進速度為0.001mm/s;
(4)將前軀體纖維置于馬弗爐中,按照2℃/min的升溫速度由室溫升至650℃,保溫2h,樣品隨爐冷卻后得到纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構,主體纖維是由sno2和zno納米顆粒復合而成,纖維直徑為0.75-1.0μm,纖維表面球狀結構是由sno2納米顆粒組成,球狀結構的直徑為1.35-1.80μm。
實施例6
(1)將0.7419g的zn(no3)2?6h2o和0.7520g的2-甲基咪唑分別溶于49ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌1h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)將0.0237g的檸檬酸氫二銨、0.0391g的十八胺和0.0781g的乙基纖維素和0.0310g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)將0.3541g的sncl4?5h2o和0.7200g的pvp加入到1.1ml的乙醇和4.4ml的dmf的混合溶劑中,攪拌得到透明溶液,然后以0.3ml/min緩慢滴加到(2)得到的溶液中,室溫攪拌10h得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為18kv,負電壓為0.5kv,接收距離為18cm,注射器推進速度為0.002mm/s;
(4)將前軀體纖維置于馬弗爐中,按照1℃/min的升溫速度由室溫升至550℃,保溫3h,樣品隨爐冷卻后得到纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構,主體纖維是由sno2和zno納米顆粒復合而成,纖維直徑為0.18-0.35μm,纖維表面球狀結構是由sno2納米顆粒組成,球狀結構的直徑為0.25-0.55μm。
對比例1
(1)將0.3541g的sncl4?5h2o和0.7000g的pvp加入到1.0ml的乙醇和4.0ml的dmf的混合溶劑中,攪拌得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為18kv,負電壓為0.5kv,接收距離為20cm,注射器推進速度為0.002mm/s;
(2)將前軀體纖維置于馬弗爐中,按照1℃/min的升溫速度由室溫升至600℃,保溫2h,樣品隨爐冷卻后得到產物。
所得產物為由sno2小顆粒組成的尺寸分布不均一的sno2微納米纖維,纖維直徑為0.20-0.80μm,團聚現象較為明顯。該產物不再是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構。由此可以看出,zif-8顆粒的引入對產品的最終形態具有重要影響。
對比例2
(1)將0.7419g的zn(no3)2?6h2o和0.1500g的2-甲基咪唑分別溶于150ml甲醇溶劑中,攪拌得到透明溶液,混合均勻后攪拌2h,經過靜置24h,用甲醇溶劑離心洗滌3次后,置于蒸發皿中自然晾干得到zif-8產物;
(2)同實施例1步驟(2);
(3)同實施例1步驟(3);
(4)同實施例1步驟(4);
所得產物為表面光滑、尺寸分布不均一的sno2/zno復合微納米纖維,纖維直徑為0.25-0.85μm。該產物不再是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構。由此可以看出,zif-8顆粒的微觀形貌對產品的最終形態具有重要影響。
對比例3
(1)同實施例1步驟(1);
(2)將0.3541g的sncl4?5h2o、0.1500g的zif-8和0.7000g的pvp加入到1.0ml的乙醇和4.0ml的dmf的混合溶劑中,攪拌得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為18kv,負電壓為0.5kv,接收距離為20cm,注射器推進速度為0.002mm/s;
(3)同實施例1步驟(4)。
所得產物為尺寸分布不均一、形貌不規則、團聚現象明顯的sno2/zno復合微納米棒,棒的直徑為0.15-2.2μm,長徑比為3.3-7.6:1。該產物不再是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構。由此可以看出,zif-8的加入量和分散穩定性對產品的最終形態具有重要影響。
對比例4
(1)同實施例1步驟(1);
(2)同實施例1步驟(2);
(3)將0.3541g的sncl4?5h2o和1.5000g的pvp加入到1.0ml的乙醇和4.0ml的dmf的混合溶劑中,攪拌得到前驅體紡絲液,通過靜電紡絲法得到前驅體纖維,紡絲參數為:正電壓為10kv,負電壓為0.5kv,接收距離為12cm,注射器推進速度為0.004mm/s;
(4)將前軀體纖維置于馬弗爐中,按照5℃/min的升溫速度由室溫升至700℃,保溫5h,樣品隨爐冷卻后得到產物。
所得產物為團聚現象明顯、尺寸分布不均一,無規則形貌的sno2/zno復合微納米顆粒,顆粒尺寸為0.35-4.5μm。該產物不再是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構。由此可以看出,靜電紡絲參數和熱處理制度的設定對產品的最終形態具有重要影響。
對比例5
(1)同實施例1步驟(1);
(2)將1.4250g的檸檬酸氫二銨、1.2980g的十八胺和2.3285g的乙基纖維素和0.3541g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)同實施例1步驟(3);
(4)同實施例1步驟(4)。
所得產物為尺寸分布不均一、形貌不規則、團聚現象明顯的sno2/zno復合微納米棒,棒的直徑為0.10-2.5μm,長徑比為4.1-7.9:1。該產物不再是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構。由此可以看出,步驟(2)中各種功能添加劑的加入量對產品的最終形態具有重要影響。
對比例6
(1)同實施例1步驟(1);
(2)將0.6920g的草酸銨、0.6875g的1-萘胺和1.2147g的硬脂酸和0.3541g的zif-8溶于2.0ml乙醇溶劑中,攪拌得到透明溶液;
(3)同實施例1步驟(3);
(4)同實施例1步驟(4)。
所得產物為形貌不規則、團聚現象明顯的sno2/zno復合微納米球,球的直徑為0.20-2.1μm。該產物不再是纖維表面具有sno2顆粒的sno2/zno復合微納米纖維結構。由此可以看出,步驟(2)中各種功能添加劑的種類對產品的最終形態具有重要影響。
- 還沒有人留言評論。精彩留言會獲得點贊!