混纖絲的制造方法、混纖絲、卷取體和織物與流程
本發明涉及混纖絲的制造方法、混纖絲、卷取體和織物。特別是涉及分散度高、適度柔軟、纖維剝離量少的混纖絲的制造方法。
背景技術:
一直以來,已知包含連續增強纖維和連續熱塑性纖維的混纖絲(也有時稱為復合纖維)(專利文獻1、專利文獻2、專利文獻3)。
例如,專利文獻1中記載了:將實質上未附著油劑或上漿劑(sizing agent)的增強復絲和作為母材的熱塑性復絲進行復合紡線時,通過在規定條件下進行處理,從而得到復合纖維的方法(專利文獻1的權利要求1等)。另外,專利文獻1中還公開了:通過對復合纖維中的熱塑性長絲進行加熱而使其增塑,與增強復絲進行半熔接或熔接的方法。
現有技術文獻
專利文獻
專利文獻1:日本特開平1-280031號公報
專利文獻2:日本特開2013-237945號公報
專利文獻3:日本特開平4-73227號公報
技術實現要素:
發明要解決的問題
包含連續增強纖維和連續樹脂纖維的混纖絲中,要求連續增強纖維和連續樹脂纖維充分分散。此處,為了提高分散度,理想的是,表面處理劑、集束劑(也有時稱為油劑、上漿劑)等處理劑少。然而,處理劑的量少時,連續增強纖維與連續樹脂纖維的密合性差,發生纖維的剝離。另外,混纖絲不是最終加工品,因此從進一步的加工適應性的觀點出發,要求適度的柔軟度。
本發明是以解決上述問題為目的而做出的,其目的在于提供維持連續增強纖維和連續樹脂纖維的高分散度、適度柔軟且纖維的剝離少的混纖絲的制造方法。另外,目的在于提供通過前述混纖絲的制造方法等得到的混纖絲。進而,目的在于提供卷取前述混纖絲而成的卷取體和使用了前述混纖絲的織物。
用于解決問題的方案
基于上述狀況,本發明人進行了深入研究,結果發現通過下述手段<1>和<8>、優選<2>~<7>和<9>~<15>能夠解決上述問題。
<1>一種混纖絲的制造方法,其包括:將表面具有熱塑性樹脂纖維處理劑的熱塑性樹脂纖維與表面具有連續增強纖維處理劑的連續增強纖維進行混纖,在構成前述熱塑性樹脂纖維的熱塑性樹脂的熔點~熔點+30K的溫度下進行加熱,
前述熱塑性樹脂的熔點與按照ASTM D 177測得的熱導率的乘積為100~150,
前述連續增強纖維處理劑的量為前述連續增強纖維的0.01~2.0重量%,
前述熱塑性樹脂纖維處理劑的量為前述熱塑性樹脂纖維的0.1~2.0重量%;其中,熔點的單位為K,熱導率的單位為W/m·K。
<2>根據<1>所述的混纖絲的制造方法,其中,前述熔點~熔點+30K的溫度下的加熱用加熱輥進行。
<3>根據<1>所述的混纖絲的制造方法,其中,前述熔點~熔點+30K的溫度下的加熱用單面加熱輥進行。
<4>根據<1>~<3>中任一項所述的混纖絲的制造方法,其中,前述熱塑性樹脂為聚酰胺樹脂和聚縮醛樹脂中的至少1種。
<5>根據<1>~<4>中任一項所述的混纖絲的制造方法,其中,前述熱塑性樹脂為由源自二胺的結構單元和源自二羧酸的結構單元構成的聚酰胺樹脂,源自二胺的結構單元的50摩爾%以上源自苯二甲胺。
<6>根據<1>~<5>中任一項所述的混纖絲的制造方法,其中,前述連續增強纖維為碳纖維或玻璃纖維。
<7>根據<1>~<6>中任一項所述的混纖絲的制造方法,其中,前述混纖絲中的前述熱塑性樹脂纖維的浸滲率為5~15%。
<8>一種混纖絲,其為包含熱塑性樹脂纖維、前述熱塑性樹脂纖維的處理劑、連續增強纖維、以及前述連續增強纖維的處理劑的混纖絲,
構成前述熱塑性樹脂纖維的熱塑性樹脂的熔點與按照ASTM D 177測得的熱導率的乘積為100~150,
前述連續增強纖維的處理劑和前述熱塑性樹脂纖維的處理劑的總量為混纖絲的0.2~4.0重量%,
將前述混纖絲進行并絲并在熔點+20℃、5分鐘、3MPa的條件下成形,在296K的水中浸漬30天后,按照ISO 527-1和ISO 527-2在23℃、卡盤間距離50mm、拉伸速度50mm/分鐘的條件下測得的拉伸強度的維持率為60~100%,
前述混纖絲的分散度為60~100%,
前述混纖絲中的前述熱塑性樹脂纖維的浸滲率為5~15%;其中,熔點的單位為K,熱導率的單位為W/m·K。
<9>根據<8>所述的混纖絲,其中,前述熱塑性樹脂為聚酰胺樹脂和聚縮醛樹脂中的至少1種。
<10>根據<8>或<9>所述的混纖絲,其中,前述熱塑性樹脂為由源自二胺的結構單元和源自二羧酸的結構單元構成的聚酰胺樹脂,源自二胺的結構單元的50摩爾%以上源自苯二甲胺。
<11>根據<10>所述的混纖絲,其中,前述源自二羧酸的結構單元的50摩爾%以上為己二酸和癸二酸中的至少一者。
<12>根據<8>~<11>中任一項所述的混纖絲,其中,前述連續增強纖維為碳纖維或玻璃纖維。
<13>根據<8>~<12>中任一項所述的混纖絲,其中,前述混纖絲為通過<1>~<7>中任一項所述的混纖絲的制造方法制造的混纖絲。
<14>一種卷取體,其是將<8>~<13>中任一項所述的混纖絲卷取成卷而成的。
<15>一種織物,其使用了<8>~<13>中任一項所述的混纖絲。
發明的效果
根據本發明,能夠提供維持連續增強纖維和連續樹脂纖維的高分散度、適度柔軟且纖維的剝離少的混纖絲的制造方法。另外,能夠提供通過前述混纖絲的制造方法等得到的混纖絲。進而,能夠提供將前述混纖絲卷取而成的卷取體和使用了前述混纖絲的織物。
附圖說明
圖1示出使用單面加熱輥加熱混纖絲的實施方式的示意圖。
圖2為示出實施例中的柔軟度測定方法中使用的基座的截面形狀的示意圖。
圖3示出實施例中的分散度測定方法中的圖像處理的一例。
具體實施方式
以下,詳細說明本發明的內容。需要說明的是,本說明書中,“~”是以包括其前后所記載的數值作為下限值及上限值的含義來使用的。
本說明書中,溫度以0℃=273K來表示。
本發明的混纖絲的制造方法的特征在于,包括:將表面具有熱塑性樹脂纖維處理劑的熱塑性樹脂纖維與表面具有連續增強纖維處理劑的連續增強纖維進行混纖,在構成前述熱塑性樹脂纖維的熱塑性樹脂的熔點~熔點+30K的溫度下進行加熱,前述熱塑性樹脂的熔點(單位:K)與按照ASTM D 177測得的熱導率(單位:W/m·K)的乘積為100~150,前述連續增強纖維處理劑的量為前述連續增強纖維的0.01~2.0重量%,前述熱塑性樹脂纖維處理劑的量為前述熱塑性樹脂纖維的0.1~2.0重量%。
通過采用這種構成,能夠提供維持連續增強纖維和連續樹脂纖維的高分散度、適度柔軟且纖維的剝離少的混纖絲的制造方法。
包含連續增強纖維和連續樹脂纖維的混纖絲中,要求連續增強纖維和連續樹脂纖維充分分散。此處,為了提高分散度,理想的是纖維的處理劑少。然而,處理劑的量少時,連續增強纖維與連續樹脂纖維的密合性差,發生纖維的剝離。本發明中,通過將處理劑的量限制為上述范圍,從而提高分散度。另一方面,處理劑的量少這一點通過將加熱溫度設為熱塑性樹脂的熔點~熔點+30K,并且對熔點與熱導率的乘積為100~150的熱塑性樹脂以上述溫度加熱來彌補。即,在這種條件下加熱時,連續樹脂纖維沒有完全浸滲于連續增強纖維,成為部分浸滲的狀態(以下在本說明書中有時稱為“微浸滲”)。通過制成這種微浸滲狀態,抑制了混纖絲中的纖維剝離。進而,對混纖絲賦予適度的柔軟度。另外,通過使連續樹脂纖維在連續增強纖維中微浸滲,從而所得到的成形品的機械強度也提高。
另一方面,熱塑性樹脂的熔點與熱導率的乘積不足100時,浸滲的推進度快,混纖絲無法成為整齊的直線狀。作為其結果,混纖絲的剛性過強,成為缺乏柔軟度的混纖絲,加工性差。特別是加工成織物、編織物等時,構成混纖絲的纖維的一部分或全部斷裂。另一方面,超過150時,浸滲變得難以推進,所得到的混纖絲過于柔軟,纖維的剝離量變多。
本發明的熱塑性樹脂的熔點與前述熱導率的乘積的下限值優選為105以上,上限值優選為140以下、更優選為135以下、進一步優選為130以下。通過設為這種范圍,更有效地發揮本發明的效果。
以下,關于本發明的混纖絲的制造方法,詳細進行說明。
<混纖>
本發明的制造方法包括:將表面具有熱塑性樹脂纖維處理劑的熱塑性樹脂纖維與表面具有連續增強纖維處理劑的連續增強纖維進行混纖的工序。混纖可以通過公知的方法進行,例如可列舉出:從表面具有熱塑性樹脂纖維處理劑的熱塑性樹脂纖維束的卷繞體和表面具有連續增強纖維處理劑的連續增強纖維束的卷繞體分別取出連續熱塑性樹脂纖維束和連續增強纖維束,一邊進行開纖一邊使連續熱塑性樹脂纖維和連續增強纖維成為一束。開纖例如可以施加吹氣來進行。
<加熱>
本發明的制造方法中,在混纖后,在構成熱塑性樹脂纖維的熱塑性樹脂的熔點~熔點+30K的溫度下進行加熱。
此處,構成熱塑性樹脂纖維的熱塑性樹脂具有2個以上熔點時,將最低的熔點作為構成熱塑性樹脂纖維的熱塑性樹脂的熔點。另外,熱塑性樹脂纖維由2種以上的熱塑性樹脂形成時,將含量最多的熱塑性樹脂的熔點作為構成熱塑性樹脂纖維的熱塑性樹脂的熔點。
加熱溫度優選為熔點+5K~熔點+30K、進一步優選為熔點+10K~熔點+30K。通過在這種范圍內加熱,能夠使熱塑性樹脂纖維微浸滲而不會完全浸滲。
作為加熱時間,沒有特別規定,例如可以設為0.5~10秒,優選為1~5秒。
對于加熱手段,沒有特別規定,可以使用公知的手段。具體而言,可例示出加熱輥、紅外線(IR)加熱器、熱風、激光照射等,優選利用加熱輥的加熱。
利用加熱輥加熱時,混纖絲成為扁平狀。通過制成扁平狀的混纖絲,加工成織物時,經絲的波紋變淺,能夠進一步提高最終得到的成形品的機械強度。
用加熱輥加熱混纖絲時,可以使用單面加熱輥進行加熱,也可以使用雙面加熱輥進行加熱。圖1為示出使用單面加熱輥進行制造的實施方式的一例的示意圖,以使混纖絲1沿著多個分開的單面加熱輥2的方式反復對混纖絲的單面進行逐一加熱。另外,使用雙面加熱輥的情況下,可以用2個加熱輥即一對加熱輥夾住混纖絲并同時加熱的雙面。本發明中,從提高生產率的觀點出發,優選使用單面加熱輥,對單面進行逐一加熱。
<其它工序>
本發明的混纖絲的制造方法中,在不超出本發明主旨的范圍內也可以包括除上述混纖和加熱以外的工序。
本發明的混纖絲的制造方法中,優選在自前述混纖工序至卷取成卷為止之間不包括其它加熱工序。另外,本發明中,也可以不使用溶劑地進行制造,因此也可以采用不包括混纖絲的干燥工序的制造方法。
本發明的混纖絲進行上述加熱后,在保持微浸滲不變的狀態下卷取成卷而制成卷取體或裝入袋中來保存。
<熱塑性樹脂纖維>
本發明的熱塑性樹脂纖維為表面具有熱塑性樹脂纖維處理劑的熱塑性樹脂纖維。
通過在熱塑性樹脂纖維的表面應用處理劑,在混纖絲的制造工序和/或之后的加工工序中能夠抑制熱塑性樹脂纖維的斷裂。特別是,熱塑性樹脂的處理劑提高熱塑性樹脂的浸滲性,即使在如上述規定的溫度條件那樣較低的溫度下加熱,也能夠達成微浸滲的狀態。
本發明中使用的連續熱塑性樹脂纖維由熱塑性樹脂組合物形成。熱塑性樹脂組合物以熱塑性樹脂作為主成分(通常組合物的90重量%以上為熱塑性樹脂),此外,適當配混有公知的添加劑等。作為本發明的實施方式的一例,熱塑性樹脂組合物中所含的樹脂可列舉出特定的1種樹脂占整體的80重量%以上的方式,進而,熱塑性樹脂組合物中所含的樹脂也可列舉出特定的1種樹脂占整體的90重量%以上的方式。
作為熱塑性樹脂,可以廣泛地使用復合材料用混纖絲中使用的物質,例如可以使用聚酰胺樹脂、聚對苯二甲酸乙二醇酯、聚對苯二甲酸丁二醇酯等聚酯樹脂、聚碳酸酯樹脂、聚縮醛樹脂等熱塑性樹脂,優選聚酰胺樹脂和聚縮醛樹脂,進一步優選聚酰胺樹脂。
關于本發明中能使用的聚酰胺樹脂和聚縮醛樹脂的詳情,在后面說明。
<<熱塑性樹脂組合物>>
本發明的連續熱塑性樹脂纖維更優選由熱塑性樹脂組合物形成。
熱塑性樹脂組合物以熱塑性樹脂作為主成分,也可以包含添加劑等。
<<<聚酰胺樹脂>>>
作為聚酰胺樹脂,可以使用公知的聚酰胺樹脂。
例如,可列舉出聚酰胺4、聚酰胺6、聚酰胺11、聚酰胺12、聚酰胺46、聚酰胺66、聚酰胺610、聚酰胺612、聚對苯二甲酰己二胺(聚酰胺6T)、聚間苯二甲酰己二胺(聚酰胺6I)、聚酰胺9T等。
另外,從成形性、耐熱性的觀點出發,更優選使用通過α,ω-直鏈脂肪族二羧酸與苯二甲胺的縮聚而得到的苯二甲胺系聚酰胺樹脂(XD系聚酰胺)。聚酰胺樹脂為2種以上的聚酰胺樹脂的混合物時,聚酰胺樹脂中的XD系聚酰胺的比率優選為50重量%以上、更優選為80重量%以上。
本發明中使用的優選的聚酰胺樹脂的一個實施方式是二胺結構單元(源自二胺的結構單元)的50摩爾%以上源自苯二甲胺的聚酰胺樹脂,且前述聚酰胺樹脂的數均分子量(Mn)為6000~30000。本實施方式的聚酰胺樹脂更優選其0.5~5重量%是重均分子量為1000以下的聚酰胺樹脂。
本發明中使用的聚酰胺樹脂如上所述優選為二胺的50摩爾%以上源自苯二甲胺且與二羧酸縮聚而成的苯二甲胺系聚酰胺樹脂。更優選為如下的苯二甲胺系聚酰胺樹脂:二胺結構單元的70摩爾%以上、進一步優選80摩爾%以上源自間苯二甲胺和/或對苯二甲胺,且二羧酸結構單元(源自二羧酸的結構單元)的優選50摩爾%以上、更優選70摩爾%以上、特別是80摩爾%以上源自碳原子數優選為4~20的α,ω-直鏈脂肪族二羧酸。
本發明中特別優選為二胺結構單元的70摩爾%以上源自間苯二甲胺且二羧酸結構單元的50摩爾%以上源自α,ω-直鏈脂肪族二羧酸的聚酰胺樹脂,進一步優選為前述二胺結構單元的70摩爾%以上源自間苯二甲胺且二羧酸結構單元的50摩爾%以上源自癸二酸的聚酰胺樹脂。
作為能用作苯二甲胺系聚酰胺樹脂的原料二胺成分的除間苯二甲胺和對苯二甲胺以外的二胺,可例示出四亞甲基二胺、五亞甲基二胺、2-甲基戊二胺、六亞甲基二胺、七亞甲基二胺、八亞甲基二胺、九亞甲基二胺、十亞甲基二胺、十二亞甲基二胺、2,2,4-三甲基六亞甲基二胺、2,4,4-三甲基六亞甲基二胺等脂肪族二胺、1,3-雙(氨基甲基)環已烷、1,4-雙(氨基甲基)環已烷、1,3-二氨基環已烷、1,4-二氨基環已烷、雙(4-氨基環己基)甲烷、2,2-雙(4-氨基環己基)丙烷、雙(氨基甲基)十氫萘、雙(氨基甲基)三環癸烷等脂環式二胺、雙(4-氨基苯基)醚、對苯二胺、雙(氨基甲基)萘等具有芳香環的二胺等,可以使用1種或混合使用2種以上。
作為二胺成分,使用苯二甲胺以外的二胺時,以二胺結構單元的50摩爾%以下、優選30摩爾%以下、更優選1~25摩爾%、特別優選5~20摩爾%的比例使用。
作為優選用作聚酰胺樹脂的原料二羧酸成分的碳原子數4~20的α,ω-直鏈脂肪族二羧酸,例如可例示出琥珀酸、戊二酸、庚二酸、辛二酸、壬二酸、己二酸、癸二酸、十一烷二酸、十二烷二酸等脂肪族二羧酸,可以使用1種或混合使用2種以上,這些之中,從聚酰胺樹脂的熔點成為適于成形加工的范圍出發,優選己二酸或癸二酸、特別優選癸二酸。
作為除上述碳原子數4~20的α,ω-直鏈脂肪族二羧酸以外的二羧酸成分,可例示出間苯二甲酸、對苯二甲酸、鄰苯二甲酸等苯二甲酸化合物、1,2-萘二羧酸、1,3-萘二羧酸、1,4-萘二羧酸、1,5-萘二羧酸、1,6-萘二羧酸、1,7-萘二羧酸、1,8-萘二羧酸、2,3-萘二羧酸、2,6-萘二羧酸、2,7-萘二羧酸之類的異構體等萘二羧酸等,可以使用1種或混合使用2種以上。
作為二羧酸成分,使用除碳原子數4~20的α,ω-直鏈脂肪族二羧酸以外的二羧酸時,從成形加工性、阻隔性的觀點出發,優選使用對苯二甲酸、間苯二甲酸。配混對苯二甲酸、間苯二甲酸時,配混比例優選為二羧酸結構單元的30摩爾%以下、更優選為1~30摩爾%、特別優選為5~20摩爾%的范圍。
進而,除二胺成分、二羧酸成分以外,作為構成聚酰胺樹脂的成分,在不損害本發明效果的范圍內也可以使用ε-己內酰胺、月桂內酰胺等內酰胺類、氨基己酸、氨基十一烷酸等脂肪族氨基羧酸類作為共聚成分。
作為聚酰胺樹脂,優選聚己二酰間苯二甲胺樹脂、聚癸二酰間苯二甲胺樹脂、聚癸二酰對苯二甲胺樹脂、及使間苯二甲胺與對苯二甲胺的混合苯二甲胺與己二酸縮聚而成的聚己二酰間苯二甲胺/對苯二甲胺混合樹脂,更優選為聚癸二酰間苯二甲胺樹脂、聚癸二酰對苯二甲胺樹脂、及使間苯二甲胺與對苯二甲胺的混合苯二甲胺與癸二酸縮聚而成的聚癸二酰間苯二甲胺/對苯二甲胺混合樹脂。這些聚酰胺樹脂存在成形加工性特別良好的傾向。
本發明中使用的聚酰胺樹脂的數均分子量(Mn)優選為6000~30000,更優選其中的0.5~5重量%是重均分子量為1000以下的聚酰胺樹脂。
將數均分子量(Mn)設為6000~30000的范圍內時,存在所得到的復合材料或其成形品的強度進一步提高的傾向。更優選的數均分子量(Mn)為8000~28000、進一步優選為9000~26000、更進一步優選為10000~24000、特別優選為11000~22000、更特別優選為12000~20000。為這種范圍時,耐熱性、彈性模量、尺寸穩定性、成形加工性變得更良好。
需要說明的是,此處所說的數均分子量(Mn)是根據聚酰胺樹脂的末端氨基濃度[NH2](μ當量/g)和末端羧基濃度[COOH](μ當量/g)由下述式算出的。
數均分子量(Mn)=2000000/([COOH]+[NH2])
另外,聚酰胺樹脂優選含有0.5~5重量%的重均分子量(Mw)為1000以下的成分。通過以這種范圍含有這種低分子量成分,所得到的聚酰胺樹脂向連續增強纖維中的浸滲性提高,因此其成形品的強度、低翹曲性變得良好。超過5重量%時,該低分子量成分滲出,強度惡化,表面外觀變差。
重均分子量為1000以下的成分的更優選含量為0.6~5重量%。
重均分子量為1000以下的低分子量成分的含量的調整可以調節聚酰胺樹脂聚合時的溫度、壓力、二胺的滴加速度等熔融聚合條件來進行。特別是可以在熔融聚合后期將反應裝置內減壓來去除低分子量成分,調節為任意的比例。另外,也可以對通過熔融聚合制造的聚酰胺樹脂進行熱水提取來去除低分子量成分,也可以在熔融聚合后進一步在減壓下進行固相聚合來去除低分子量成分。固相聚合時,可以調節溫度、減壓度而將低分子量成分控制為任意的含量。另外,也可以通過將重均分子量為1000以下的低分子量成分在之后添加到聚酰胺樹脂中來進行調節。
需要說明的是,重均分子量1000以下的成分量的測定可以使用東曹株式會社(TOSOH CORPORATION)制造的“HLC-8320GPC”,由通過凝膠滲透色譜(GPC)測定得到的標準聚甲基丙烯酸甲酯(PMMA)換算值求出。需要說明的是,可以在作為測定用柱使用2根“TSKgel SuperHM-H”、溶劑使用三氟乙酸鈉濃度為10mmol/l的六氟異丙醇(HFIP)、樹脂濃度為0.02重量%、柱溫為40℃(313K)、流速為0.3ml/分鐘、折射率檢測器(RI)的條件下測定。另外,標準曲線是將6級的PMMA溶解于HFIP并測定而制作的。
本發明中使用的聚酰胺樹脂的分子量分布(重均分子量/數均分子量(Mw/Mn))優選為1.8~3.1。分子量分布更優選為1.9~3.0、進一步優選為2.0~2.9。通過將分子量分布設為這種范圍,存在容易得到機械物性優異的復合材料的傾向。
聚酰胺樹脂的分子量分布例如可以通過適當選擇聚合時使用的引發劑、催化劑的種類、量及反應溫度、壓力、時間等聚合反應條件等來調整。另外,通過將由不同聚合條件得到的平均分子量不同的多種聚酰胺樹脂混合、或使聚合后的聚酰胺樹脂分別沉淀,從而也可以進行調整。
分子量分布可以通過GPC測定求出,具體而言,可以在作為裝置使用東曹株式會社制造的“HLC-8320GPC”、作為柱使用2根東曹株式會社制造的“TSK gel Super HM-H”、洗脫液三氟乙酸鈉濃度為10mmol/l的六氟異丙醇(HFIP)、樹脂濃度為0.02重量%、柱溫為40℃(313K)、流速為0.3ml/分鐘、折射率檢測器(RI)的條件下進行測定,以標準聚甲基丙烯酸甲酯換算的值的形式求出。另外,標準曲線是將6級的PMMA溶解于HFIP并測定而制作的。
另外,關于聚酰胺樹脂的熔融粘度,在聚酰胺樹脂的熔點(Tm)+30℃(Tm+303K)、剪切速度為122秒-1、聚酰胺樹脂的含水率為0.06重量%以下的條件下測定時,優選為50~1200Pa·s。通過將熔融粘度設為這種范圍,容易將聚酰胺樹脂加工成薄膜或纖維。需要說明的是,后述那樣的聚酰胺樹脂具有2個以上熔點時,將高溫側的吸熱峰的峰值溫度作為熔點來進行測定。
熔融粘度的更優選范圍為60~500Pa·s、進一步優選為70~100Pa·s。
聚酰胺樹脂的熔融粘度例如可以通過適當選擇原料二羧酸成分和二胺成分的投料比、聚合催化劑、分子量調節劑、聚合溫度、聚合時間來調整。
另外,聚酰胺樹脂的吸水時的彎曲模量保持率優選為85%以上。通過將吸水時的彎曲模量保持率設為這種范圍,成形品的高溫高濕度下的物性降低少,存在翹曲等形狀變化少的傾向。
此處,吸水時的彎曲模量保持率被定義為由聚酰胺樹脂形成的彎曲試驗片的吸水0.5重量%時的彎曲模量相對于吸水0.1重量%時的彎曲模量的比率(%),該比率高意味著即使吸濕也不易使彎曲模量降低。
吸水時的彎曲模量保持率更優選為90%以上、進一步優選為95%以上。
聚酰胺樹脂的吸水時的彎曲模量保持率例如可以通過對苯二甲胺與間苯二甲胺的混合比例來控制,對苯二甲胺的比例越多,彎曲模量保持率越良好。另外,通過控制彎曲試驗片的結晶度,也能進行調整。
關于聚酰胺樹脂的吸水率,以在23℃下在水中浸漬1星期后取出并擦除水分后立即測定時的吸水率計優選為1重量%以下、更優選為0.6重量%以下、進一步優選為0.4重量%以下。為該范圍時,容易防止成形品的由吸水導致的變形,此外,抑制加熱加壓時等的將復合材料成形加工時的發泡,能夠得到氣泡少的成形品。
另外,聚酰胺樹脂可以適宜地使用末端氨基濃度([NH2])優選不足100μ當量/g、更優選為5~75μ當量/g、進一步優選為10~60μ當量/g、且末端羧基濃度([COOH])優選不足150μ當量/g、更優選為10~120μ當量/g、進一步優選為10~100μ當量/g的物質。通過使用這種末端基團濃度的聚酰胺樹脂,將聚酰胺樹脂加工成薄膜狀或纖維狀時粘度容易穩定,此外,存在與后述碳二亞胺化合物的反應性變得良好的傾向。
另外,末端氨基濃度相對于末端羧基濃度之比([NH2]/[COOH])優選為0.7以下、更優選為0.6以下、特別優選為0.5以下。該比大于0.7時,有時在聚合聚酰胺樹脂時難以控制分子量。
末端氨基濃度可以將聚酰胺樹脂0.5g在20~30℃下攪拌溶解于30ml的苯酚/甲醇(4:1)混合溶液并用0.01N的鹽酸滴定來測定。另外,末端羧基濃度可以如下算出:將聚酰胺樹脂0.1g在200℃下溶解于30ml的苯甲醇,在160℃~165℃的范圍內加入酚紅溶液0.1ml。對于該溶液用使0.132g的KOH溶解于苯甲醇200ml而得到的滴定液(以KOH濃度計0.01mol/l)進行滴定,顏色變化為黃~紅,將顏色變化消失的時刻作為終點,從而算出。
本發明的聚酰胺樹脂中,反應所得二胺單元相對于反應所得二羧酸單元的摩爾比(反應所得二胺單元的摩爾數/反應所得二羧酸單元的摩爾數,以下有時稱為“反應摩爾比”)優選為0.97~1.02。通過設為這種范圍,變得容易將聚酰胺樹脂的分子量、分子量分布控制在任意的范圍。
反應摩爾比更優選不足1.0、進一步優選不足0.995、特別是不足0.990,下限更優選為0.975以上、進一步優選為0.98以上。
此處,反應摩爾比(r)由下述式求出。
r=(1-cN-b(C-N))/(1-cC+a(C-N))
式中,
a:M1/2
b:M2/2
c:18.015(水的分子量(g/mol))
M1:二胺的分子量(g/mol)
M2:二羧酸的分子量(g/mol)
N:末端氨基濃度(當量/g)
C:末端羧基濃度(當量/g)
需要說明的是,作為二胺成分、二羧酸成分,由分子量不同的單體合成聚酰胺樹脂時,自不待言,M1和M2根據作為原料配混的單體的配混比(摩爾比)來計算。需要說明的是,合成釜內為完全的封閉體系時,投入的單體的摩爾比與反應摩爾比一致,但實際合成裝置不會成為完全封閉體系,因此投料的摩爾比與反應摩爾比不一定一致。投入的單體也不一定完全反應,因此投料的摩爾比與反應摩爾比不一定一致。因此,反應摩爾比是指由成品的聚酰胺樹脂的末端基團濃度求出的實際上反應了的單體的摩爾比。
聚酰胺樹脂的反應摩爾比的調整可以通過將原料二羧酸成分和二胺成分的投料摩爾比、反應時間、反應溫度、苯二甲胺的滴加速度、釜內的壓力、減壓開始時機等反應條件設為適當值來進行。
聚酰胺樹脂的制造方法為所謂的鹽法時,為了使反應摩爾比為0.97~1.02,具體而言例如將原料二胺成分/原料二羧酸成分比設定為該范圍,使反應充分推進即可。另外,向熔融二羧酸中連續滴加二胺的方法的情況下,除了將投料比設為該范圍之外,也可以在滴加二胺的過程中控制回流的二胺量,將所滴加的二胺去除至反應體系外。具體而言,通過將回流塔的溫度控制在最佳范圍、將填充塔的填充物、所謂拉西環、萊辛環、鞍環等控制為適當的形狀、填充量,從而將二胺去除至體系外即可。另外,通過縮短二胺滴加后的反應時間,也能夠將未反應的二胺去除至體系外。進而,通過控制二胺的滴加速度,也能夠將未反應的二胺根據需要去除至反應體系外。通過這些方法,即使投料比偏離期望范圍,也能將反應摩爾比控制在規定的范圍。
聚酰胺樹脂的制造方法沒有特別限定,利用現有公知的方法、聚合條件制造。聚酰胺樹脂的縮聚時,也可以加入少量的單胺、單羧酸作為分子量調節劑。例如,通過將由包含苯二甲胺的二胺成分與己二酸、癸二酸等二羧酸形成的鹽在水的存在下以加壓狀態升溫,一邊去除添加的水及縮合水一邊在熔融狀態下聚合的方法來制造。另外,也可以通過將苯二甲胺直接加入至熔融狀態的二羧酸中并在常壓下縮聚的方法來制造。此時,為了使反應體系保持均勻的液狀狀態,將二胺連續地加入至二羧酸中,在此期間以反應溫度不會低于所生成的低聚酰胺及聚酰胺的熔點的方式將反應體系升溫并推進縮聚。
另外,聚酰胺樹脂在通過熔融聚合法制造后可以進行固相聚合。固相聚合的方法沒有特別限定,利用現有公知的方法、聚合條件來制造。
本發明中,聚酰胺樹脂的熔點優選為150~310℃、更優選為180~300℃。
另外,聚酰胺樹脂的玻璃化轉變溫度優選為50~100℃、更優選為55~100℃、特別優選為60~100℃。為該范圍時,存在耐熱性變得良好的傾向。
需要說明的是,熔點是指通過DSC(差示掃描量熱測定)法觀測的升溫時的吸熱峰的峰值溫度。另外,玻璃化轉變溫度是指,將試樣暫時加熱熔融來消除由熱歷程對結晶性造成的影響后再次升溫而測定的玻璃化轉變溫度。測定可以使用例如島津制作所株式會社(SHIMADZU CORPORATION)制造的“DSC-60”,在試樣量設為約5mg、作為氣氛氣體使氮氣以30ml/分鐘流通、升溫速度為10℃/分鐘的條件下從室溫加熱至預想的熔點以上的溫度使其熔融,利用此時觀測的吸熱峰的峰值溫度求出熔點。接著,將熔融的聚酰胺樹脂用干冰驟冷,以10℃/分鐘的速度再次升溫至熔點以上的溫度,可以求出玻璃化轉變溫度。
本發明中使用的聚酰胺樹脂中也可以包含除上述苯二甲胺系聚酰胺樹脂以外的其它聚酰胺樹脂。作為其它聚酰胺樹脂,可列舉出聚酰胺66、聚酰胺6、聚酰胺46、聚酰胺6/66、聚酰胺10、聚酰胺612、聚酰胺11、聚酰胺12、由六亞甲基二胺、己二酸和對苯二甲酸形成的聚酰胺66/6T、由六亞甲基二胺、間苯二甲酸和對苯二甲酸形成的聚酰胺6I/6T等。它們的配混量優選為聚酰胺樹脂成分的5重量%以下、更優選為1重量%以下。
<<<聚縮醛樹脂>>>
聚縮醛樹脂只要包含二價的氧亞甲基作為結構單元就沒有特別限定,可以為僅包含二價的氧亞甲基作為結構單元的均聚物,也可以為包含二價的氧亞甲基和碳數為2以上的二價的氧亞烷基作為結構單元的共聚物。
二價的氧亞烷基的碳數通常為2~6。作為碳數為2~6的氧亞烷基,例如可列舉出氧亞乙基、氧亞丙基、氧亞丁基、氧亞戊基及氧亞己基等。
聚縮醛樹脂中,碳數2以上的氧亞烷基在氧亞甲基和碳數2以上的氧亞烷基的總重量中所占的比例沒有特別限定,例如為0~30重量%即可。
為了制造上述聚縮醛樹脂,通常使用三氧雜環己烷作為主原料。另外,為了向聚縮醛樹脂中導入碳數2以上的氧亞烷基,可以使用例如環狀甲縮醛、環狀醚。作為環狀甲縮醛的具體例,例如可列舉出1,3-二氧雜環戊烷、1,3-二噁烷、1,3-二氧雜環庚烷、1,3-二氧雜環辛烷、1,3,5-三氧雜環己烷、1,3,6-三氧雜環辛烷等,作為環狀醚的具體例,例如可列舉出環氧乙烷、環氧丙烷和環氧丁烷等。為了向聚縮醛樹脂中導入氧亞乙基,例如使用1,3-二氧雜環戊烷即可,為了導入氧亞丙基,使用1,3-二噁烷即可,為了導入氧亞丁基,導入1,3-二氧雜環庚烷即可。
<<<彈性體>>>
本發明中使用的熱塑性樹脂組合物也可以包含彈性體成分。
作為彈性體成分,例如可以使用聚烯烴系彈性體、二烯系彈性體、聚苯乙烯系彈性體、聚酰胺系彈性體、聚酯系彈性體、聚氨酯系彈性體、氟系彈性體、有機硅系彈性體等公知的彈性體,優選為聚烯烴系彈性體及聚苯乙烯系彈性體。作為這些彈性體,為了賦予對于聚酰胺樹脂的相容性,在自由基引發劑的存在下或不存在下利用α,β-不飽和羧酸及其酸酐、丙烯酰胺以及它們的衍生物等改性而得到的改性彈性體。
彈性體成分的含量為熱塑性樹脂組合物中的通常30重量%以下、優選為20重量%以下、特別是10重量%以下。
另外,上述熱塑性樹脂組合物也可以將一種或多種熱塑性樹脂共混來使用。
進而,在不損害本發明的目的/效果的范圍內,可以在本發明中使用的熱塑性樹脂組合物中加入抗氧化劑、熱穩定劑等穩定劑、耐水解性改良劑、耐候穩定劑、消光劑、紫外線吸收劑、成核劑、增塑劑、分散劑、阻燃劑、抗靜電劑、防著色劑、抗膠凝劑、著色劑、脫模劑等添加劑等。它們的詳情可以參考日本特許第4894982號公報第0130~0155段的記載,將這些內容并入到本說明書中。
<<連續熱塑性樹脂纖維的處理劑>>
本發明的熱塑性樹脂纖維在表面具有熱塑性樹脂的處理劑。本發明的熱塑性樹脂纖維的處理劑的量通常為熱塑性樹脂纖維的0.1~2.0重量%。下限值優選為0.5重量%以上、更優選為0.8重量%以上。作為上限值,優選為1.8重量%以下、更優選為1.5重量%以下。通過設為這種范圍,連續熱塑性樹脂纖維的分散變得良好,容易得到更均質的混纖絲。另外,制造混纖絲時連續熱塑性樹脂纖維產生與機械的摩擦力、纖維彼此的摩擦力,此時連續熱塑性樹脂纖維有時斷裂,但通過設為上述范圍,能夠更有效地防止纖維的切斷。另外,為了得到均質的混纖絲,對連續熱塑性樹脂纖維施加機械應力,能夠更有效地防止由此時的應力導致連續熱塑性樹脂纖維切斷。
處理劑只要具有將連續熱塑性樹脂纖維集束的功能,則其種類沒有特別規定。作為處理劑,可例示出礦物油和動/植物油等油劑、非離子表面活性劑、陰離子表面活性劑和兩性表面活性劑等表面活性劑。
更具體而言,優選為酯系化合物、亞烷基二醇系化合物、聚烯烴系化合物、苯基醚系化合物、聚醚系化合物、有機硅系化合物、聚乙二醇系化合物、酰胺系化合物、磺酸鹽系化合物、磷酸鹽系化合物、羧酸鹽系化合物和它們中的2種以上的組合。
另外,處理劑的量采用通過后述實施例中說明的方法而測得的值。
<<連續熱塑性樹脂纖維的基于處理劑的處理方法>>
連續熱塑性樹脂纖維的基于處理劑的處理方法只要能夠達成期望目的就沒有特別規定。例如,可列舉出:對連續熱塑性樹脂纖維賦予將處理劑溶解于溶液而得到的物質,使處理劑附著于連續熱塑性樹脂纖維的表面。或者,也可以將處理劑吹氣到連續熱塑性樹脂纖維的表面上。
<<連續熱塑性樹脂纖維的形態>>
本發明中使用的連續熱塑性樹脂纖維通常為多個纖維形成為束狀而成的連續熱塑性樹脂纖維束,使用連續熱塑性樹脂纖維束制造本發明的混纖絲。
本發明的連續熱塑性樹脂纖維是指具有超過6mm的纖維長度的熱塑性樹脂纖維。本發明中使用的連續熱塑性樹脂纖維的平均纖維長度沒有特別限制,從使成形加工性良好的觀點出發,優選為1~20000m的范圍、更優選為100~10000m、進一步優選為1000~7000m。
本發明中使用的連續熱塑性樹脂纖維通常使用連續熱塑性樹脂纖維形成為束狀而成的連續熱塑性樹脂纖維束來制造,所述連續熱塑性樹脂纖維束平均1條的總纖度優選為40~600dtex、更優選為50~500dtex、進一步優選為100~400dtex。通過設為這種范圍,所得到的混纖絲中的連續熱塑性樹脂纖維的分散狀態變得良好。構成所述連續熱塑性樹脂纖維束的纖維數優選為1~200f、更優選為5~100f、進一步優選為10~80f、特別優選為20~50f。通過設為這種范圍,所得到的混纖絲中的連續熱塑性樹脂纖維的分散狀態變得更良好。
本發明中,為了制造1條混纖絲,優選以1~100條的范圍使用上述連續熱塑性樹脂纖維束,更優選以10~80條的范圍使用,進一步優選以20~50條的范圍使用。通過設為這種范圍,更有效地發揮本發明的效果。
用于制造1條混纖絲的上述連續熱塑性樹脂纖維的總纖度優選為200~12000dtex、更優選為1000~10000dtex。通過設為這種范圍,更有效地發揮本發明的效果。
用于制造1條混纖絲的上述連續熱塑性樹脂纖維的總纖維數優選為10~10000f、更優選為100~5000f、進一步優選為500~3000f。通過設為這種范圍,混纖絲的混纖性提高,能夠得到作為復合材料的物性和質感更優異的混纖絲。進而,通過將纖維數設為10f以上,經開纖的纖維變得更容易均勻混合。另外,設為10000f以下時,不易形成任一纖維偏重存在的區域,得到更具均勻性的混纖絲。
本發明中使用的連續熱塑性樹脂纖維束的拉伸強度優選為2~10gf/d。通過設為這種范圍,存在更容易制造混纖絲的傾向。
<連續增強纖維>
本發明的連續增強纖維為表面具有連續增強纖維處理劑的連續增強纖維。
通過對連續增強纖維的表面應用處理劑,連續增強纖維的處理劑提高已熔融的熱塑性樹脂與連續增強纖維的密合性,抑制纖維的剝離。
作為連續增強纖維,可列舉出碳纖維、玻璃纖維、植物纖維(包括洋麻(Kenaf)、竹纖維等)、氧化鋁纖維、硼纖維、陶瓷纖維、金屬纖維(鋼纖維等)等無機纖維;芳綸纖維、聚氧亞甲基纖維、芳香族聚酰胺纖維、聚對苯撐苯并二噁唑纖維、超高分子量聚乙烯纖維等有機纖維等。優選為無機纖維,其中,由于具有輕量且高強度、高彈性模量的優異特征,因此優選使用碳纖維或玻璃纖維,進一步優選為碳纖維。碳纖維可以優選地使用聚丙烯腈系碳纖維、瀝青系碳纖維。另外,也可以使用木質素、纖維素等植物來源原料的碳纖維。通過使用碳纖維,存在所得到的成形品的機械強度進一步提高的傾向。
<<連續增強纖維的處理劑>>
本發明的連續增強纖維在表面具有連續增強纖維的處理劑。本發明的連續增強纖維的處理劑的量通常為連續增強纖維的0.01重量%~2.0重量%。作為下限值,優選為0.1重量%以上、更優選為0.3重量%以上。作為上限值,優選為1.5重量%以下、更優選為1.3重量%以下。
處理劑的量采用通過后述實施例中說明的方法而測得的值。
作為本發明中使用的連續增強纖維的處理劑,優選采用日本特許第4894982號公報的第0093和0094段記載的物質,這些內容并入到本說明書中。
具體而言,本發明中使用的處理劑優選為環氧樹脂、氨基甲酸酯樹脂、硅烷偶聯劑、水不溶性聚酰胺樹脂和水溶性聚酰胺樹脂中的至少1種,更優選為環氧樹脂、氨基甲酸酯樹脂、水不溶性聚酰胺樹脂和水溶性聚酰胺樹脂中的至少1種,進一步優選為水溶性聚酰胺樹脂。
作為環氧樹脂,可列舉出環氧烷烴、烷烴二環氧化物、雙酚A-縮水甘油醚、雙酚A-縮水甘油醚的二聚體、雙酚A-縮水甘油醚的三聚體、雙酚A-縮水甘油醚的低聚物、雙酚A-縮水甘油醚的聚合物、雙酚F-縮水甘油醚、雙酚F-縮水甘油醚的二聚體、雙酚F-縮水甘油醚的三聚體、雙酚F-縮水甘油醚的低聚物、雙酚F-縮水甘油醚的聚合物、硬脂基縮水甘油醚、苯基縮水甘油醚、環氧乙烷月桂醇縮水甘油醚、乙二醇二縮水甘油醚、聚乙二醇二縮水甘油醚、丙二醇二縮水甘油醚等縮水甘油基化合物;苯甲酸縮水甘油酯、對甲基苯甲酸縮水甘油酯、硬脂酸縮水甘油酯、月桂酸縮水甘油酯、棕櫚酸縮水甘油酯、油酸縮水甘油酯、亞油酸縮水甘油酯、亞麻酸縮水甘油酯、鄰苯二甲酸二縮水甘油酯等縮水甘油酯化合物;四縮水甘油基氨基二苯基甲烷、三縮水甘油基氨基苯酚、二縮水甘油基苯胺、二縮水甘油基甲苯胺、四縮水甘油基間苯二甲胺、三縮水甘油基氰脲酸酯、三縮水甘油基異氰脲酸酯等縮水甘油胺化合物。
作為氨基甲酸酯樹脂,例如可以使用使多元醇、由油脂與多元醇發生酯交換而得到的多元醇及多異氰酸酯反應而得到的氨基甲酸酯樹脂。
作為上述多異氰酸酯,例如可列舉出1,4-四亞甲基二異氰酸酯、1,6-六亞甲基二異氰酸酯、2,2,4-三甲基六亞甲基二異氰酸酯、2,8-二異氰酸酯甲基己酸酯等脂肪族異氰酸酯類;3-異氰酸酯甲基-3,5,5-三甲基環己基異氰酸酯、甲基環己基-2,4-二異氰酸酯等的脂環族二異氰酸酯類;甲代亞苯基二異氰酸酯、二苯基甲烷二異氰酸酯、1,5-環烷二異氰酸酯、二苯基甲基甲烷二異氰酸酯、四烷基二苯基甲烷二異氰酸酯、4,4-二芐基二異氰酸酯、1,3-亞苯基二異氰酸酯等芳香族二異氰酸酯類;氯化二異氰酸酯類、溴化二異氰酸酯類等,它們可以單獨使用或以2種以上的混合物的形式使用。
作為前述多元醇,可列舉出通常氨基甲酸酯樹脂的制造中使用的各種多元醇,例如二乙二醇、丁二醇、己二醇、新戊二醇、雙酚A、環已烷二甲醇、三羥甲基丙烷、甘油、季戊四醇、聚乙二醇、聚丙二醇、聚酯多元醇、聚己內酯、聚四亞甲基醚二醇、聚硫醚多元醇、聚縮醛多元醇、聚丁二烯多元醇、呋喃二甲醇等,它們可以單獨使用或以2種以上的混合物的形式使用。
作為硅烷偶聯劑,例如可列舉出氨基丙基三乙氧基硅烷、苯基氨基丙基三甲氧基硅烷、縮水甘油基丙基三乙氧基硅烷、甲基丙烯酰氧基丙基三甲氧基硅烷、乙烯基三乙氧基硅烷等三烷氧基或三烯丙氧基硅烷化合物、脲基硅烷、硫醚硅烷、乙烯基硅烷、咪唑硅烷等。
此處,水不溶性聚酰胺樹脂是指,在25℃下將1g聚酰胺樹脂添加于100g水中時,99重量%以上不溶解。
使用水不溶性聚酰胺樹脂時,優選在水或有機溶劑中使粉末狀的水不溶性聚酰胺樹脂分散或懸濁來使用。可以在這種粉末狀的水不溶性聚酰胺樹脂的分散物或懸濁液中浸漬混纖維束來使用,使其干燥而制成混纖絲。
作為水不溶性聚酰胺樹脂,可列舉出聚酰胺樹脂6、聚酰胺樹脂66、聚酰胺樹脂610、聚酰胺樹脂11、聚酰胺樹脂12、苯二甲胺系聚酰胺樹脂(優選為聚己二酰苯二甲胺、聚癸二酰苯二甲胺)、以及對于它們的共聚物的粉體添加非離子系、陽離子系、陰離子系或作為它們的混合物的表面活性劑進行乳化分散而得到的物質。水不溶性聚酰胺樹脂的市售品例如以水不溶性聚酰胺樹脂乳液的形式銷售,例如可列舉出住友精化株式會社制造的SEPOLSION PA、Michaelman制造的Michem Emulsion。
此處,水溶性聚酰胺樹脂是指,在25℃下將1g聚酰胺樹脂添加于100g水中時,其99重量%以上溶于水。
作為水溶性聚酰胺樹脂,可列舉出丙烯酸接枝化N-甲氧基甲基化聚酰胺樹脂、賦予了酰胺基的N-甲氧基甲基化聚酰胺樹脂等改性聚酰胺。作為水溶性聚酰胺樹脂,例如可列舉出東麗株式會社制造的AQ-聚酰胺樹脂、Nagase ChemteX Corporation制造的TORESIN等市售品。
處理劑可以僅使用1種,也可以使用2種以上。
本發明中,通過將連續熱塑性樹脂纖維與連續增強纖維用少量的處理劑進行混纖,能夠提高混纖絲中的連續增強纖維的分散度。
<<連續增強纖維的基于處理劑的處理方法>>
連續增強纖維的基于處理劑的處理方法可以采用公知的方法。例如,可列舉出:將連續增強纖維浸漬于包含處理劑的液體(例如,水溶液),使處理劑附著于連續增強纖維的表面。另外,也可以將處理劑吹氣至連續增強纖維的表面。進而,也可以使用用處理劑進行了處理的連續增強纖維的市售品,也可以在將市售品的處理劑洗掉后再次以期望量重新處理。
<<連續增強纖維的形態>>
連續增強纖維是指,具有超過6mm的纖維長度的連續增強纖維。本發明中使用的連續增強纖維的平均纖維長度沒有特別限制,從使成形加工性良好的觀點出發,優選為1~20000m的范圍、更優選為100~10000m、進一步優選為1000~7000m。
本發明中使用的連續增強纖維的平均1條混纖絲的總纖度優選為100~50000dtex、更優選為500~40000dtex、進一步優選為1000~10000dtex、特別優選為1000~3000dex。通過設為這種范圍,加工變得更容易,所得到的混纖絲的彈性模量/強度變得更優異。
本發明中使用的連續增強纖維的平均1條混纖絲的總纖維數優選為500~50000f、更優選為500~20000f、進一步優選為1000~10000f、特別優選為1500~5000f。通過設為這種范圍,混纖絲中的連續增強纖維的分散狀態變得更良好。
1條混纖絲中,為了使連續增強纖維滿足規定的總纖度和總纖維數,可以利用1條連續增強纖維束來制造,也可以使用多條連續增強纖維束來制造。本發明中,優選使用1~10條連續增強纖維束來制造,更優選使用1~3條連續增強纖維束來制造,進一步優選使用1條連續增強纖維束來制造。
本發明的混纖絲中所含的連續增強纖維的平均拉伸模量優選為50~1000GPa、更優選為200~700GPa。通過設為這種范圍,混纖絲整體的拉伸模量變得更良好。
<混纖絲>
本發明的混纖絲的特征在于,其為包含熱塑性樹脂纖維、前述熱塑性樹脂纖維的處理劑、連續增強纖維、以及前述連續增強纖維的處理劑的混纖絲,構成前述熱塑性樹脂纖維的熱塑性樹脂的熔點(單位:K)與按照ASTM D 177測得的熱導率(W/m·K)的乘積為100~150,前述連續增強纖維的處理劑和前述熱塑性樹脂纖維的處理劑的總量為混纖絲的0.2~4.0重量%,將前述混纖絲進行并絲并在熔點+20℃、5分鐘、3MPa的條件下成形,在296K的水中浸漬30天后,按照ISO 527-1和ISO 527-2在23℃、卡盤間距離50mm、拉伸速度50mm/分鐘的條件下測得的拉伸強度的維持率(本說明書中有時也稱為“吸濕時強度維持率”)為60~100%,前述混纖絲的分散度為60~100%,前述混纖絲中的前述熱塑性樹脂纖維的浸滲率為5~15%。
通過制成這種混纖絲,得到適度柔軟且纖維的剝離量少的混纖絲。
本發明的混纖絲中的熱塑性樹脂纖維、熱塑性樹脂纖維的處理劑、連續增強纖維、連續增強纖維的處理劑分別與混纖絲的制造方法中說明的含義相同,優選范圍也相同。
本發明的混纖絲中的處理劑的總量通常為混纖絲的0.2~4.0重量%。作為下限值,優選為0.8重量%以上、更優選為1.0重量%以上。作為上限值,優選為3.5重量%以下、更優選為2.8重量%以下。
連續增強纖維的處理劑和前述熱塑性樹脂纖維的處理劑的總量采用按照后述實施例的混纖絲的處理劑的量而測得的量。
需要說明的是,本發明的混纖絲中的處理劑的主旨也包括其一部分或全部與其它表面處理劑、熱塑性樹脂等混纖絲中的其它成分發生了反應的情況。
關于熱塑性樹脂的熔點(單位:K)與熱導率(單位:W/m·K)的乘積,與混纖絲的制造方法中說明的含義相同,優選范圍也相同。
本發明的混纖絲的上述吸濕時強度維持率通常為60~100%。吸濕時強度保持率優選為70~100%、進一步優選為75~100%。
本發明的混纖絲中的連續熱塑性樹脂纖維和連續增強纖維的分散度通常為60~100%、優選為70~100%。通過設為這種范圍,混纖絲顯示出更均勻的物性,進而使成形時間縮短,成形品的外觀進一步提高。另外,使用其制作成形品時得到機械物性更優異的成形品。
本發明的分散度是指,表示在混纖絲中連續熱塑性樹脂纖維和連續增強纖維以何種程度均勻分散的指標,采用通過后述實施例中示出的方法測定的值。另外,關于超深度彩色3D形狀測定顯微鏡,在實施例中說明的設備停產、難以獲取的情況下,采用同種設備測得的值。
分散度越大,意味著連續熱塑性樹脂纖維和連續增強纖維更均勻地分散。
本發明的混纖絲中的熱塑性樹脂纖維的浸滲率通常為5~15%、優選為5~12%、更優選為5~10%。通過制成這種微浸滲的狀態,能夠制成適度柔軟且纖維的剝離少的混纖絲。浸滲率采用通過后述實施例中說明的方法測得的值。
進而,本發明的混纖絲中,也可以包含除上述熱塑性樹脂纖維、熱塑性樹脂纖維的處理劑、連續增強纖維、連續增強纖維的處理劑以外的其它成分,具體而言,可例示出短纖維長度碳纖維、碳納米管、富勒烯、微纖維素纖維、滑石、云母等。這些其它成分的配混量優選為混纖絲的5重量%以下。
另外,本發明的混纖絲只要是使用處理劑將連續熱塑性樹脂纖維與連續增強纖維制成束狀而成的,則其形狀沒有特別規定,包括截面為扁平狀、圓形等各種形狀的混纖絲。本發明的混纖絲優選為扁平狀。此處,扁平狀是指,凹凸少、基本上平整。
一條混纖絲的制造中使用的連續熱塑性樹脂纖維的纖度的總和與連續增強纖維的纖度的總和之比(連續熱塑性樹脂纖維的纖度的總和/連續增強纖維的纖度的總和)優選為0.1~10、更優選為0.1~6.0、進一步優選為0.8~2.0。
一條混纖絲的制造中使用的纖維數的總和(將連續熱塑性樹脂纖維的纖維數的總和與連續增強纖維的纖維數的總和求和而得的纖維數)優選為10~100000f、更優選為100~100000f、進一步優選為200~70000f、更進一步優選為300~20000f、特別優選為400~10000f、特別優選為500~5000f。通過設為這種范圍,混纖絲的混纖性提高,得到作為復合材料的物性和質感更優異的混纖絲。另外,任一纖維偏重存在的區域少,各纖維彼此容易更均勻地分散。
一條混纖絲的制造中使用的連續熱塑性樹脂纖維的纖維數的總和與連續增強纖維的纖維數的總和之比(連續熱塑性樹脂纖維的纖維數的總和/連續增強纖維的纖維數的總和)優選為0.001~1、更優選為0.001~0.5、進一步優選為0.05~0.2。通過設為這種范圍,混纖絲的混纖性提高,得到作為復合材料的物性和質感更優異的混纖絲。另外,混纖絲中的連續熱塑性樹脂纖維和連續增強纖維優選各纖維彼此更均勻地分散,若為上述范圍,則各纖維彼此容易更均勻地分散。
本發明的混纖絲的制造方法沒有特別規定,例如可以通過上述本發明的混纖絲的制造方法來制造。
<混纖絲的用途>
本發明的混纖絲在通過上述本發明的混纖絲的制造方法而制造后,可以保持微浸滲的狀態不變地卷取成卷而制成卷取體,或者進一步加工成各種成形材料。作為使用了混纖絲的成形材料,可例示出織物、編結物、編織帶、無紡布、無序氈、編織物等。本發明的混纖絲適度柔軟且纖維的剝離少,因此織物、編織物優異,特別是織物優異。
作為編織帶的形態,沒有特別限制,可例示出方結、扁結、圓結等。
作為織物的形態,沒有特別限制,可以為平紋、八經緞紋、四經緞紋、斜紋等中的任意種類。另外,也可以為所謂斜織(bias weave)。進而,也可以為如日本特開昭55-30974號公報中記載那樣實質上不具有彎曲的所謂無卷曲織物。
織物的情況下,可例示出經絲和緯絲中至少一者為本發明的混纖絲的方式。經絲和緯絲中的另一者可以設為本發明的混纖絲,根據期望的特性,也可以為增強纖維、熱塑性樹脂纖維。作為經絲和緯絲中的另一者使用熱塑性樹脂纖維的情況的一個形態,可例示出使用以與構成本發明的混纖絲的熱塑性樹脂相同的熱塑性樹脂作為主成分的纖維。
作為編織物的形態,沒有特別限制,可以自由選擇經編、緯編、拉舍爾編織等公知的編織方式。
作為無紡布的形態,沒有特別限制,例如可以將本發明的混纖絲切斷,形成絨毛(fleece),將混纖絲之間結合,制成無紡布。絨毛的形成可以使用干式法、濕式法等。另外,混纖絲間的結合可以采用化學鍵合法、熱接合法等。
另外,也可以以如下的形態來使用:使本發明的混纖絲沿一個方向并絲而成的帶狀或片狀的基材、編織帶、繩狀的基材、或將這些基材層疊2張以上而得到的層疊物。
進而,也優選以將本發明的混纖絲、編織帶、織物、編織物或無紡布等層疊并加熱加工而成的復合材料的形態來使用。加熱加工例如可以在熱塑性樹脂的熔點+10~30℃的溫度下進行。
使用了本發明的混纖絲、成形材料或復合材料的成形品例如可以適宜地用于個人電腦、OA設備、AV設備、手機等電氣/電子設備、光學設備、精密設備、玩具、家用及辦公電器產品等的部件、外殼、進而汽車、航空器、船舶等的部件。特別適合于制造具有凹部、凸部的成形品。
實施例
以下列舉實施例來進一步具體說明本發明。以下的實施例中示出的材料、用量、比例、處理內容、處理步驟等可以在不超出本發明的主旨的范圍內適當變更。因此,本發明的范圍不限定于以下示出的具體例子。需要說明的是,本實施例中的各種性能評價在沒有特別說明的情況下在23℃、相對濕度50%的氣氛下進行。
<聚酰胺樹脂MPXD10的合成例>
將癸二酸在氮氣氣氛下的反應釜內加熱熔化后,一邊攪拌內容物,一邊在加壓(0.35MPa)下緩慢滴加對苯二甲胺(三菱瓦斯化學株式會社制造)與間苯二甲胺(三菱瓦斯化學株式會社制造)的摩爾比為3:7的混合二胺,使得二胺與癸二酸(伊藤制油(Itoh Oil Chemicals Co.)制造、產品名Sebacic Acid TA)的摩爾比約為1:1,一邊將溫度上升至235℃。滴加結束后,繼續反應60分鐘,調整分子量為1000以下的成分量。反應結束后,將內容物以線料狀取出,用造粒機粒料化,得到聚酰胺(MPXD10)。以下稱為“MPXD10”。
<聚酰胺樹脂MXD10的合成例>
在反應釜內將癸二酸(伊藤制油(Itoh Oil Chemicals Co.)制造、產品名Sebacic Acid TA)在170℃下加熱熔融后,一邊攪拌內容物,一邊在加壓(0.4MPa)下緩慢滴加間苯二甲胺(三菱瓦斯化學株式會社制造),使得與癸二酸的摩爾比約為1:1,一邊將溫度上升至210℃。滴加結束后,減壓至0.078MPa并繼續反應30分鐘,調整分子量為1000以下的成分量。反應結束后,將內容物以線料狀取出,用造粒機粒料化,得到聚酰胺(MXD10)。以下稱為“MXD10”。
<聚酰胺樹脂PXD10的合成例>
向具備攪拌機、分餾器、冷卻器、溫度計、滴加裝置及氮氣導入管、線料模頭的內容積50升的反應容器中稱量并投入精密稱量的癸二酸(伊藤制油制造、Sebacic Acid TA)8950g(44.25mol)、次磷酸鈣12.54g(0.074mol)、醋酸鈉6.45g(0.079mol)。將反應容器內充分氮氣置換后,用氮氣加壓至0.4MPa,一邊攪拌一邊從20℃升溫至190℃,用55分鐘使癸二酸均勻熔融。接著,在攪拌下用110分鐘滴加對苯二甲胺(三菱瓦斯化學株式會社制造)5960g(43.76mol)。在此期間,反應容器的內部溫度連續上升至293℃。滴加工序中將壓力控制為0.42MPa,生成水通過分餾器及冷卻器去除至體系外。分餾器的溫度控制在145~147℃的范圍。對苯二甲胺滴加結束后,在反應容器內壓力0.42MPa下繼續20分鐘縮聚反應。在此期間,反應容器的內部溫度上升至296℃。然后,用30分鐘將反應容器內壓力從0.42MPa減壓至0.12MPa。在此期間,內部溫度升溫至298℃。然后以0.002MPa/分鐘的速度減壓,用20分鐘減壓至0.08MPa,調整分子量為1000以下的成分量。減壓完成時的反應容器內的溫度為301℃。然后,將體系內用氮氣加壓,在反應容器內的溫度301℃、樹脂溫度301℃下從線料模頭將聚合物以線料狀取出,用20℃的冷卻水冷卻,將其粒料化,得到約13kg的聚酰胺樹脂。需要說明的是,冷卻水中的冷卻時間為5秒,線料的拉取速度為100m/分鐘。以下稱為“PXD10”。
<其它樹脂>
MXD6:己二酰間苯二甲胺樹脂(三菱瓦斯化學株式會社制造、等級S6007)
PA66:聚酰胺樹脂66(東麗株式會社制造、AMILAN CM3001)
POM:聚縮醛樹脂(三菱工程塑料株式會社制造、型號:F20-03)
PEEK:聚醚醚酮樹脂(VICTREX公司制造、450G)
PPS:聚苯硫醚樹脂(寶理塑料株式會社制造、0220A9)
PS:聚苯乙烯樹脂(出光興產株式會社制造、XAREC)
<增強纖維>
CF:碳纖維、東麗株式會社制造、使用用環氧樹脂進行了表面處理的碳纖維
GF:玻璃纖維、Nitto Boseki Co.,Ltd.制造、使用用硅烷偶聯劑進行了表面處理的玻璃纖維
<熱塑性樹脂的纖維化>
上述熱塑性樹脂通過以下的手法制成纖維狀。
使用具有30mmφ的螺桿的單螺桿擠出機將熱塑性樹脂熔融擠出,從60孔的模頭以線料狀擠出,一邊卷取成卷一邊拉伸,得到卷取為卷繞體的熱塑性樹脂纖維束。關于熔融溫度,聚酰胺樹脂(PXD10)為300℃、其它聚酰胺樹脂為280℃、POM樹脂為210℃、PEEK樹脂為380℃、PPS樹脂為340℃、PS樹脂為300℃。
<樹脂纖維的處理劑>
聚氧乙烯氫化蓖麻油(花王株式會社制造、EMANON 1112)
<熱塑性樹脂纖維的表面處理>
上述樹脂纖維的處理劑通過以下的手法涂布于熱塑性樹脂纖維。
將樹脂纖維的處理劑(油劑)裝滿深型桶,將表面經橡膠處理的輥以輥的下部分與油劑接觸的方式設置,通過使輥旋轉,制成油劑一直附著于輥表面的狀態。通過使樹脂纖維與該輥接觸,從而在樹脂纖維的表面涂布油劑。
<實施例1~6和比較例1~9的混纖絲的制造>
從卷繞體分別拉出連續熱塑性樹脂纖維和連續增強纖維,通過多個引導件,施加吹氣進行開纖。一邊開纖,一邊將連續熱塑性樹脂纖維和連續增強纖維制成一束,進而通過多個引導件,施加吹氣進行均勻化,從而進行混纖。然后,在表中記載的加熱溫度下,使纖維束沿著表面經Teflon(注冊商標)處理的單面加熱輥對單面進行3秒加熱,接著將混纖絲的反面同樣地處理,得到混纖絲。所使用的加熱輥由Kaji Group co.ltd.制造的加熱器(DCD4028-1)和圓筒(DCD4014A)(外徑100mm)構成。其中,關于表中示為“未加熱”的比較例,沒有進行加熱。
<處理劑的量的測定>
<<連續增強纖維>>
將經表面處理的連續增強纖維5g(設為重量(X))浸漬于甲乙酮200g,將處理劑在25℃下溶解,進行清洗。減壓下加熱至60℃使甲乙酮蒸發,回收殘渣,測量其重量(Y)。處理劑的量由Y/X(重量%)算出。對于樹脂纖維,也通過同樣的方法測定處理劑的量。
<<混纖絲>>
將混纖絲5g(設為重量(X))浸漬于甲乙酮200g,將處理劑在25℃下溶解,進行超聲波清洗。減壓下加熱至60℃使甲乙酮蒸發,回收殘渣,測量其重量(Y)。處理劑的量由Y/X(重量%)算出。
<分散度的測定>
如下進行觀察來測定混纖絲的分散度。
切取混纖絲,用環氧樹脂包埋,對相當于混纖絲的截面部的面進行研磨,使用超深度彩色3D形狀測定顯微鏡VK-9500(控制部)/VK-9510(測定部)(Keyence制造)拍攝截面圖。如圖3所示,拍攝圖像中,呈放射狀以等間隔畫6條輔助線,測量位于各輔助線上的連續增強纖維區域的長度記作a1、a2、a3···ai(i=n)。同時測量位于各輔助線上的熱塑性樹脂纖維區域的長度記作b1、b2、b3···bi(i=m)。由下述式算出分散度。
<浸滲率的測定>
切取混纖絲,用環氧樹脂包埋,對相當于混纖絲的截面部的面進行研磨,使用超深度彩色3D形狀測定顯微鏡VK-9500(控制部)/VK-9510(測定部)(Keyence制造)拍攝截面圖。用數字式顯微鏡觀察所制作的成形品的截面。對于所得到的截面照片,使用圖像分析軟件ImageJ選擇連續增強纖維的浸滲有熱塑性樹脂的區域,測定其面積。浸滲率以連續增強纖維的浸滲有熱塑性樹脂的區域/截面積(單位%)的形式表示。
<柔軟度的測定>
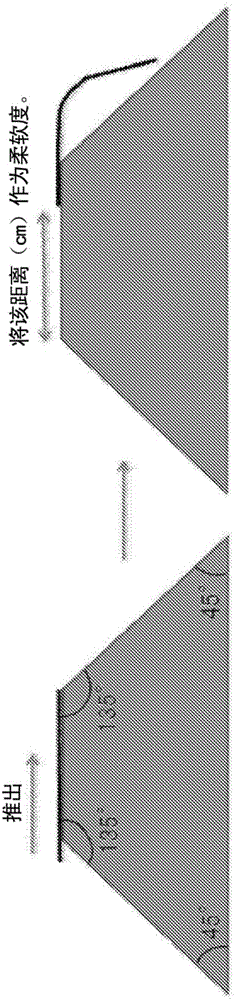
將混纖絲放置于圖2中示出截面圖那樣的具有45°斜面的截面為梯形的硬紙板制造的基座的邊緣,以0.5cm/1秒的速度推出。將從基座的上表面端部移動至碰觸斜面為止的距離(cm)作為柔軟度的指標。距離越長越柔軟。根據從基座的上表面端部移動至碰觸斜面為止的距離,如下進行區分。
A:16.0cm~18.0cm
B:15.0cm~19.0cm(其中,相當于A的情況除外)
C:不符合A、B的任一者。
<纖維剝離量的測定>
對于所得到的混纖絲,通過以下的方法測定纖維剝離量。
首先,將透明膠帶(NICHIBAN CO.,LTD.制造、CELLOTAPE 405AP、CT405AP-15、15mmx35m)切出50mm。接著,用鑷子放置在電子天平上,測定僅透明膠帶的重量。接著,將混纖絲70mm,粘貼于透明膠帶的粘接部。用指腹按壓粘接部使其密合后,在按住混纖絲的未粘接于透明膠帶的部分的狀態下剝離透明膠帶。殘留在透明膠帶上的纖維之中,將從透明膠帶突出的部分的纖維切掉。由下述式算出纖維剝離量。單位由mg/cm2表示。
((賦予混纖絲并剝離而得到的透明膠帶的重量)-(僅透明膠帶的重量))/(透明膠帶的面積)
<成形品的制造>
將上述得到的混纖絲沿一個方向排列,在構成混纖絲的熱塑性樹脂的熔點+20℃、3MPa的條件下進行5分鐘熱壓,從所得到的成形品切出1mmt×20cm×2cm的試驗片。
<拉伸強度>
對于所得到的成形品,將纖維方向作為拉伸方向,通過ISO 527-1和ISO527-2所述的方法在測定溫度23℃、卡盤間距離50mm、拉伸速度50mm/分鐘的條件下測定拉伸強度。單位由MPa表示。
<吸濕時強度維持率>
與上述同樣地測定將上述得到的成形品在296K的水中浸漬30天后的拉伸強度。如下算出吸濕時強度維持率。需要說明的是,30天水浸漬前的拉伸強度采用剛成形后的拉伸強度。
拉伸強度維持率(單位%)=(30天水浸漬后的拉伸強度)/(30天水浸漬前的拉伸強度)
<織物的制造>
通過上述熱塑性樹脂的纖維化,制造熱塑性樹脂纖維束。熱塑性樹脂纖維束使用與混纖絲中使用的熱塑性樹脂纖維相同的纖維,設為纖維數34f、纖維束的直徑110dtex。
將上述得到的混纖絲作為經絲,將熱塑性樹脂纖維束作為緯絲,使用劍桿織機進行織造。以織物的單位面積重量為240g/m2的方式調整。
<織物的成形性的評價>
對于上述織物的制造中得到的物體,如下評價。
A:得到組織整齊、沒有起毛的織物。
B:能夠制成織物,但存在起毛、或者織物中的混纖絲的一部分纖維斷裂。
C:起毛和/或松解嚴重,或者混纖絲硬而折斷,無法形成織物。
將結果示于下述表。
[表1]
[表2]
由上述明顯可知,實施例1~6的混纖絲通過制成所謂微浸滲從而在加工工序中不易發生纖維紊亂,連續纖維能夠保持直線狀,物性得以提高。
另一方面,未在規定條件下進行加熱處理的比較例1~9的纖維剝離量多或不具有適度的柔軟度,在成形為織物時,在操作方面,纖維在空氣中散亂或織物無法成形。
產業上的可利用性
本發明的混纖絲作為混纖紗(commingle yarn)這樣的新一代混纖絲而期待廣泛的利用。
附圖標記說明
1 混纖絲
2 單面加熱輥
- 還沒有人留言評論。精彩留言會獲得點贊!