一種具有界面共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化電極材料及其制備方法與流程
本發明屬于材料技術領域,特別涉及一種具有界面共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化電極材料及其制備方法。
背景技術:
聚苯胺(pani)作為電極材料,具有較高的贗電容比容量且自放電較少、成本低且具有優良的動力學性能,但載流子(離子等)在聚苯胺材料內部擴散緩慢,導致電容器的功率性能偏低且循環性能較差。活性炭纖維材料具有比表面積大、孔結構發達豐富、電導率高、化學穩定性好、柔性可彎折等特點,近年來被廣泛用于超級電容器電極材料,但其能量密度較低,成為制約其應用的一個瓶頸。
為解決二者作為電極材料的瓶頸問題,將炭纖維膜與導電pani有效結合,作為一種新型的超級電容器電極材料引起廣大科研作者的關注。其關鍵是如何實現pani/炭兩種材料間的均勻分散,且二者之間具有相當牢固和強度的連接界面,并具有納米結構的復合材料,有助于提高其作為電極材料的電化學性能。通過調控連接界面,能夠實現pani納米微纖在炭膜表面的均勻包覆及結合界面強度較高,進而制備出性能優異的超級電容器用電極材料。
zhou等(ying-kezhou,ben-linhe,wen-jiazhou,etal.electrochemicalcapacitanceofwell-coatedsingle-walledcarbonnanotubewithpolyanilinecomposites[j].electrochimicaacta,2004,49(2):257-262.)通過原位化學聚合法制備了pani/單壁cnts復合電極材料,發現cnts和pani顆粒之間形成了緊密的電荷傳輸混合體,而不是單純的弱分子連結。這種電子傳輸混合體降低了離子擴散阻抗,有利于電荷的傳輸,改善了電極的功率特性。但是該方法不適用于無定型的多孔炭。因此,要想通過構建一種“緊密的電荷傳輸混合體”來提高pani-多孔炭復合體系的電化學性能,有必要提出一種新的方法。
技術實現要素:
本發明的目的在于提供一種具有界面共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化電極材料及其制備方法,該方法制備的材料具有強的界面作用力與呈納米微纖的聚苯胺陣列,將具有骨架穩定性的炭纖維膜材料與具有較高贗電容的導電pani材料有效而牢固地結合,兩種材料之間的相互作用增強,會形成更大的π電子離域體系,使兩相間的電子傳遞也由鏈間跳躍模式轉變為鏈內的直接傳導,進而大幅度提高電極材料的性能。
為實現上述目的,本發明通過下述技術方案實現:
一種具有界面共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料的制備方法,包括以下步驟:
(1)將十六烷基三甲基溴化銨和樟腦磺酸加入到水中混合,然后加入到正己烷和正己醇的混合液中,得到混合物;將苯胺基甲基三乙氧基硅烷功能化多孔炭膜和苯胺分散至上述的混合物中,在室溫下攪拌均勻,形成a溶液;其中,十六烷基三甲基溴化銨、樟腦磺酸、水、正己烷和正己醇的混合液、苯胺基甲基三乙氧基硅烷功能化多孔炭膜、苯胺的比為(1.8~2.2)g:(3~3.5)g:(4~6)ml:(25~35)ml:(0.25~0.5)g:(1~2)ml;
(2)將十六烷基三甲基溴化銨和過硫酸銨加入到水中,然后添加至正己烷和正己醇的混合液中,形成b溶液;其中,十六烷基三甲基溴化銨、過硫酸銨、水、正己烷和正己醇的混合液的比為(1.8~2.2)g:(1.3~1.7)g:(4~6)ml:(25~35)ml;
(3)將b溶液滴加到a溶液中,攪拌均勻后,在0~5℃,靜態條件下聚合后過濾沉淀物,得到氮磷共摻雜多孔炭膜@聚苯胺雜化材料;其中,樟腦磺酸與過硫酸銨的比為(3~3.5)g:(1.3~1.7)g。
本發明進一步的改進在于,步驟(1)中攪拌的速度為200~400r/min,時間為4h。
本發明進一步的改進在于,步驟(1)和步驟(2)中正己烷和正己醇的體積比為5:1。
本發明進一步的改進在于,步驟(3)中以1~2滴/秒的速度進行滴加,聚合時間為23-25h。
本發明進一步的改進在于,步驟(1)中黑色nd42功能化多孔炭膜通過以下過程制得:
(1)將聚丙烯腈、三苯基膦和分子篩sba-15在加熱下,溶于n,n-二甲基甲酰胺中,混合均勻,得到復合紡絲液;通過靜電紡絲獲得納米纖維膜,再通過預氧化、炭化、co2活化及刻蝕,獲得氮磷共摻雜多孔炭膜;其中,聚丙烯腈、三苯基膦、sba-15的比為(2~4)g:(1~2)g:(1~2)g,復合紡絲液中聚丙烯腈質量含量為7~12%;
(2)將硝酸水溶液和氮磷共摻雜活性炭膜混合后加入到容器中,攪拌回流5小時后趁熱過濾,并用蒸餾水洗滌至中性,干燥,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm;其中,硝酸水溶液中硝酸與氮磷共摻雜活性炭膜的質量比為(15~20)g:(0.5~1)g;
(3)將上述所得hno3-npcm分散在甲苯與苯胺基甲基三乙氧基硅烷的混合液中,超聲分散,然后回流反應24h;之后冷卻、抽濾,采用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色,干燥,冷卻得到黑色nd42功能化多孔炭膜;其中,hno3-npcm:甲苯:苯胺基甲基三乙氧基硅烷的比為(0.3~0.8)g:(80~120)ml:(3~8)ml。
本發明進一步的改進在于,靜電紡絲的條件為:轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,采用的針頭尺寸為φ0.06mm,電壓為25kv,轉速為750r/min。
本發明進一步的改進在于,預氧化的具體過程為:在空氣氣氛下,以1℃/min自室溫升至280℃恒溫2h;炭化的具體過程為:在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h。
本發明進一步的改進在于,co2活化的具體過程為:炭化結束后將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降溫至室溫;刻蝕的具體過程為:利用質量濃度20%的氫氟酸將co2活化的材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,干燥,得氮磷共摻雜多孔炭膜。
本發明進一步的改進在于,硝酸水溶液質量濃度15~20%。
一種具有界面共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料,其特征在于,該材料在酸性電解液中比電容為394~549.5f/g。
本發明相對于現有技術,具有如下的有益效果:
本發明通過將苯胺基甲基三乙氧基硅烷功能化多孔炭膜和苯胺分散到含有十六烷基三甲基溴化銨和樟腦磺酸的溶液中,使樟腦磺酸摻雜的pani原位接枝在多孔炭纖維膜的表面,形成具有共價鍵鏈接的牢固均勻包覆層。由于有界面共價鍵的存在,將具有骨架穩定性的多孔炭纖維與具有較高贗電容的導電pani材料有效而牢固地結合,兩種材料之間的相互作用增強,會形成更大的π電子離域體系,使兩相間的電子傳遞也由鏈間跳躍模式轉變為鏈內的直接傳導,進而大幅度提高電極材料的比電容及循環穩定性。
當該氮磷共摻雜多孔炭膜@聚苯胺雜化材料用于超級電容器電極材料時,在酸性電解液中獲得的比電容為394~549.5f/g,相對于單一氮磷共摻雜多孔炭膜(281.1f/g)、聚苯胺(372.5f/g)以及無共價鍵鏈接的pani-ac復合材料(374.8f/g),本發明所制備出具有共價鍵結合的氮磷共摻雜多孔炭膜@聚苯胺雜化材料,作為電極材料比電容數值顯著提高;且充放電過程中曲線的形狀變形較小,更接近矩形,說明電化學性能更接近理想的電容行為,具有優異的循環穩定性。所以本發明具有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料可用作優秀的超級電容器電極材料。
進一步的,本發明采用靜電紡絲法制備出的炭納米纖維膜,經苯胺基甲基三乙氧基硅烷功能化處理后,通過苯胺單體在其表面原位聚合后,可獲得聚苯胺以微纖均勻分布在功能炭膜表面且具有共價鍵結合的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
進一步的,多孔炭纖維膜經過硝酸溶液處理后,炭纖維的表面含有豐富的羧基和羥基,可以與nd42水解后的羥基縮合,使偶聯劑以化學鍵接枝在炭材料的表面。另外,nd42分子上的活性苯胺基團又可以和苯胺單體進行共聚反應;采用含苯胺基團的硅烷偶聯劑苯胺基甲基三乙氧基硅烷,對經過硝酸活化后的多孔炭進行表面功能化處理,然后以苯胺單體在功能化多孔炭表面原位共聚,抽濾洗滌至中性,干燥后得到超級電容器用具有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
附圖說明
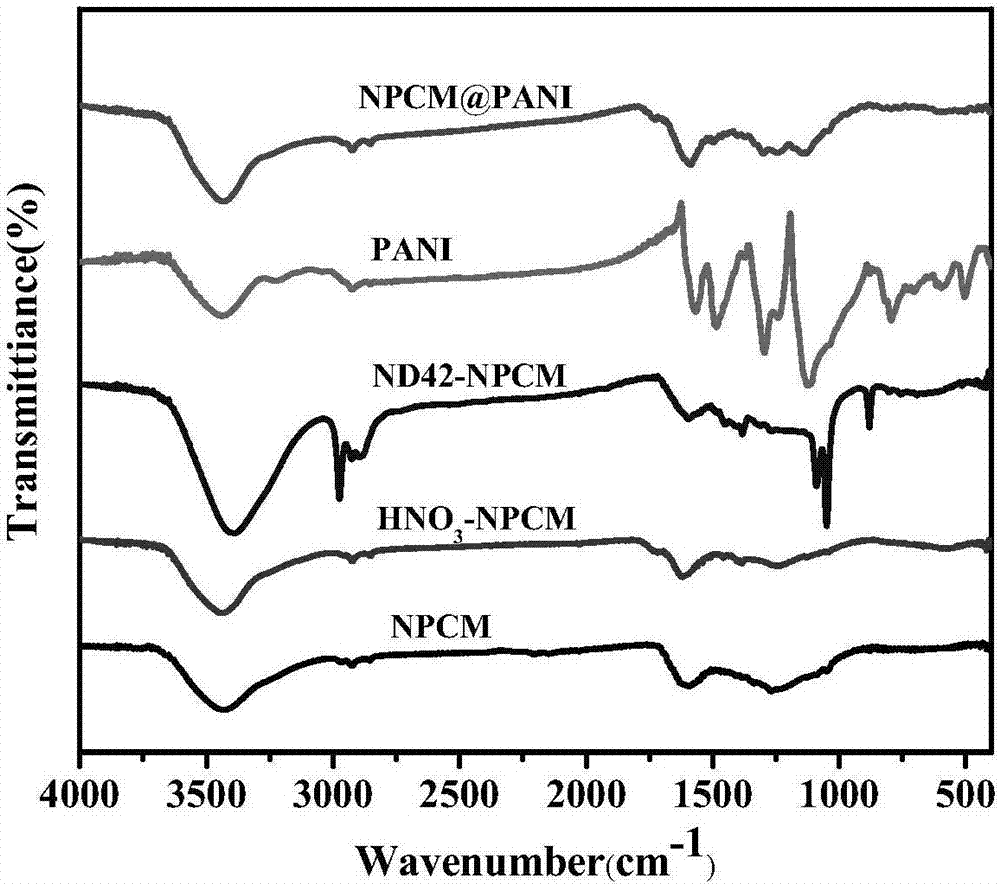
圖1實施例1所得氮磷共摻雜多孔炭膜@聚苯胺雜化材料及對比例1~2中所得氮磷共摻雜多孔炭膜、hno3-npcm、nd42-npcm、pani等5個試樣的紅外曲線。
圖2為實施例1所得氮磷共摻雜多孔炭膜@聚苯胺雜化材料的放大5000倍微觀形貌照片。
圖3為實施例1所得氮磷共摻雜多孔炭膜@聚苯胺雜化材料的放大20000倍微觀形貌照片。
圖4為實施例1制得的具有共價鍵氮磷共摻雜多孔炭膜@聚苯胺雜化材料及其它對比試樣的充放電曲線,在電流密度為1a/g時所測得。
圖5為實施例1制得的有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料循環伏安曲線圖。
圖6為實施例1制得的有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料恒流充放電曲線圖。
圖7為實施例1制得的有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料及無共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺復合材料在不同電流密度下的比電容值。
圖8為具有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料合成原理示意圖。
具體實施方式
下面結合實施例及附圖對本發明作進一步詳細的描述,但本發明的實施方式不限于此。
本發明以工業活性炭為基材,先經硝酸對其活化處理,后用含苯胺基團的偶聯劑nd42對其進行功能化處理,再將苯胺在其表面原位聚合,獲得具有界面共價鍵鏈接的pani-ac復合材料,具體包括以下步驟:
(1)將(2~4)克聚丙烯腈、(1~2)克三苯基膦和(1~2)克分子篩sba-15在70℃油浴下,溶于n,n-二甲基甲酰胺(dmf)中,配置成聚丙烯腈含量為(7-12)%的混合紡絲液。用磁力攪拌器連續攪拌8h,使混合體系充分溶解并混合均勻,得到復合紡絲液。通過靜電紡絲法(轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,20號針頭,針頭尺寸為φ0.06mm,電壓25kv,轉速750r/min)獲得納米纖維膜。再將該膜經預氧化、炭化、活化及刻蝕等步驟獲得氮磷共摻雜多孔炭膜。
其中,預氧化:在空氣氣氛下,以1℃/min從室溫升至280℃,恒溫2h;
炭化:在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h;
co2活化:炭化結束后將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降溫至室溫。
刻蝕:利用20%的hf酸將上述活化炭材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,在干燥箱內110℃下干燥過夜得氮磷共摻雜多孔炭膜,記為npcm。
(2)按硝酸水溶液濃度(15~20)%,硝酸與npcm的質量比為(15~20)g:(0.5~1)g。,混合加入到三口燒瓶中,50℃水浴下恒溫磁力攪拌回流5小時;待反應完全后趁熱過濾,并用蒸餾水洗滌至中性,濾出物在110℃烘箱中干燥12h,冷卻后得到hno3-npcm。
(3)按hno3-npcm:甲苯:nd42的用量按(0.3~0.8)g:(80~120)ml:(3~8)ml稱量,將hno3-npcm在超聲作用下分散在甲苯與nd42偶聯劑的混合液中,超聲分散0.5h,加熱至110℃下回流反應24h,反應結束后冷卻、抽濾,用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色,將產物在50℃的真空烘箱中干燥24h,冷卻后待用,得到nd42-npcm。
(4)首先將正己烷與正己醇按5:1(體積比)配成混合溶劑(正己烷/正己醇);將(1.8~2.2)g十六烷基三甲基溴化銨和(3~3.5)g樟腦磺酸混合溶于(4~6)ml去離子水中,然后加入到(25~35)ml的混合溶劑(正己烷/正己醇)中,將(0.25~0.5)g的多孔炭膜和(1~2)ml苯胺分散至上述的混合體系中,同時在室溫下200-400r/min下攪拌4個小時,標記為a溶液。通過相似的過程,b溶液是由(1.8~2.2)g十六烷基三甲基溴化銨和(1.3~1.7)g過硫酸銨溶于(4~6)ml去離子水中,并添加(25~35)ml的混合溶劑(正己烷/正己醇)中而配制成的。隨后,兩個系統都在冰水浴中200-400r/min攪拌預冷至0~5℃,然后將b溶液以1~2滴/秒的速度加入到a溶液中。200-400r/min下攪拌5分鐘后,整個混合物在大約0~5℃,靜態條件下聚合24小時。過濾沉淀物,緊接著分別用去離子水、乙醇和丙酮進行清洗,最后在60℃下真空干燥24個小時,得到乳液法制備的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
其中,a溶液中,ctab:csa:水:(正己烷/正己醇):nd42-npcm:an=(1.8~2.2)g:(3~3.5)g:(4~6)ml:(25~35)ml:(0.25~0.5)g:(1~2)ml。
b溶液中,ctab:aps:水:(正己烷/正己醇)=(1.8~2.2)g:(1.3~1.7)g:(4~6)ml:(25~35)ml。
實施例1
一種具有界面共價鍵鏈接的聚苯胺-多孔炭復合材料的制備方法,包括以下步驟:
(1)稱量聚丙烯腈:三苯基膦:sba-15的質量比為2克:1克:1克,以21克n,n-二甲基甲酰胺為溶劑,在70℃油浴下,將聚丙烯腈、三苯基膦以及sba-15溶于n,n-二甲基甲酰胺(dmf)中,用磁力攪拌器以300r/min連續攪拌8h,使混合體系充分溶解并混合均勻,然后用200w的超聲波超聲0.5h,得到聚丙烯腈質量含量為8%的混合紡絲液。通過靜電紡絲(轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,20號針頭,針頭尺寸為φ0.06mm,電壓25kv,轉速750r/min)獲得納米纖維膜。再通過預氧化、炭化、活化及刻蝕,等獲得氮磷共摻雜多孔炭膜。
預氧化:在空氣氣氛下,以1℃/min升至280℃恒溫2h;
炭化:在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h;
co2活化:炭化結束后將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降至室溫。
刻蝕:利用質量分數20%的氫氟酸將上述活化炭材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,在干燥箱內110℃下干燥12h,得氮磷共摻雜多孔炭膜,記為npcm。
(2)稱取硝酸并配制成質量濃度16%的硝酸水溶液100ml,將其和0.6克氮磷共摻雜活性炭膜混合后加入到三口燒瓶中,50℃水浴下恒溫磁力攪拌回流5小時。待反應完全后趁熱過濾,并用蒸餾水洗滌至中性,濾出物在110℃烘箱中干燥12h,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm。
(3)取0.4克上述所得hno3-npcm分散在100ml甲苯與4ml苯胺基甲基三乙氧基硅烷即nd42的混合液中,用玻璃棒將黑色硝酸活化多孔炭膜壓入液面以下并200w下超聲分散0.5h;然后加熱至110℃下回流反應24h,50r/min磁力攪拌;之后冷卻、抽濾,用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色;將產物在50℃的真空烘箱中干燥24h,冷卻得到黑色nd42功能化多孔炭膜,記作nd42-npcm。
(4)首先將正己烷與正己醇按5:1(體積比)配成混合溶劑(正己烷/正己醇);將1.98g十六烷基三甲基溴化銨和3.3g樟腦磺酸混合溶于5ml去離子水中,然后加入到30ml的混合溶劑(正己烷/正己醇)中,將0.3g的多孔炭膜和1.2ml苯胺分散至上述的混合體系中,并在室溫下攪拌4個小時,標記為a溶液。
通過相似的過程,b溶液是由1.98g十六烷基三甲基溴化銨和1.5g過硫酸銨溶于5ml去離子水中,并添加30ml的混合溶劑(正己烷/正己醇)中而配制成的。
隨后,將a溶液和b溶液都在冰水浴中攪拌預冷至0~5℃,然后將b溶液以1滴/秒的速度加入到a溶液中。以為300r/min磁力攪拌5分鐘后,整個混合物在大約0~5℃,靜態條件下聚合24小時。過濾沉淀物,緊接著分別用去離子水、乙醇和丙酮進行清洗,最后在60℃下真空干燥24個小時,得到乳液法制備的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
(5)為了檢測制備所得具有共價鍵鏈接的npcm@pani雜化材料電化學性能,將制備得到的npcm@pani作電極材料,以三電極體系進行測試。測試用的電解液為1mol/l的h2so4溶液。
對比例1
不加入苯胺單體,僅用實施例1中步驟(1)所得的npcm。
對比例2
不加入活性炭,僅用實施例1中步驟(4)所得的聚苯胺。
對比例3
僅用實施例1中步驟(1)所得的npcm,不經步驟(2)、(3)的處理,直接通過步驟(4)在npcm上聚合沉積pani,獲得無共價鍵的npcm/pani復合材料。
參見表1,表1為實施例1與對比例1~3所得試樣的比電容值,從表1可以看出具有界面共價鍵鏈接的npcm@pani雜化材料比電容值(549.5f/g)最高,既優于單一氮磷共摻雜多孔炭膜(281.1f/g)、聚苯胺(372.5f/g),也優于無共價鍵鏈接的pani-ac復合材料(374.8f/g)。
表1實施例1與對比例1~3所得試樣的比電容值(1a/g)
圖1為實施例1中所得npcm@pani雜化材料與對比例1~2中所得的氮磷共摻雜多孔炭膜、hno3-npcm、nd42-npcm、pani等5個試樣的紅外曲線圖。從圖中可看出,所有試樣在3400~3450cm-1都有一個明顯的液態水吸收峰,這是由于烘干不充分所致。多孔炭膜在1250cm-1處出現的一個寬大的吸收峰,可能是多孔炭膜中酚羥基的c-o振動吸收峰,在1600cm-1處出現的吸收峰可歸屬于-coo的反對稱伸縮,這是由于炭膜是經過co2活化后導致的,恰好可以提高該炭膜的親水性。苯胺甲基三乙氧基硅烷處理后的活性炭膜(nd42-npcm),其在1734cm-1、1573cm-1、1211cm-1出現了吸收峰,相對于多孔炭膜而言,nd42-npcm的碳鏈中c-n的伸縮振動吸收峰在1160~1000cm-1范圍內,苯環上c-n的伸縮振動吸收峰在1350~1250cm-1范圍內,在1000~1350cm-1處出現了一個寬泛的吸收峰,對應于c-o在1100cm-1及1350~1260cm-1處的伸縮振動峰、si-o-c及si-o-si(鏈狀)在1100~1000cm-1處振動、仲胺中的c-n在1350~1280cm-1處振動的聯合吸收峰,且在2965cm-1處出現了一個明顯的吸收峰,該峰克歸屬于苯胺甲基三乙氧基硅烷偶聯劑中的-ch3反對稱伸縮。可以推斷出在1350~1000cm-1之間處吸收峰明顯增強是由于c-o、c-n、si-o共同作用的結果,由此說明苯胺甲基三乙氧基硅烷處理活性炭膜的方法是可行的,苯胺甲基三乙氧基硅烷偶聯劑可以通過這種方法以化學鍵的方式接枝在活性炭的表面。雜化材料npcm@pani的曲線與純pani較為相似,只是由于nd42-npcm的存在,部分吸收峰有所減弱。這說明由于nd42-npcm的存在,影響了聚苯胺中原子的振動頻率,而聚苯胺具有一種全共軛的分子架構,這種影響會隨著大π鍵而影響整個分子鏈的振動頻率,導致樣品紅外光譜的特征吸收峰發生移動。以上的變化說明,在nd42-npcm的表面發生了苯胺的聚合反應,且二者之間產生了一定的物理化學作用,形成了具有共價鍵鏈接的復合材料。
圖2及圖3分別為實施例1所得氮磷共摻雜多孔炭膜@聚苯胺雜化材料的放大5000倍及20000倍的微觀形貌照片。可見每個炭纖維表面都均勻生長著聚苯胺短纖;從放大圖中可清晰地看出聚苯胺短纖垂直于多孔炭纖維表面并有一定的孔隙存在。
圖4為實施例1制得的具有共價鍵氮磷共摻雜多孔炭膜@聚苯胺雜化材料及其它對比試樣的充放電曲線,在電流密度為1a/g時所測得。曲線1為氮磷共摻雜多孔炭膜的充放電曲線,近似對稱的三角形,具有典型的雙電層電容特性。曲線2(pani)電極的充放電曲線在0.1~0.6v間有明顯的氧化還原峰,體現出了pani的法拉第贗電容特性;而在0.6~0.8v間則表現為具有對稱三角形的雙電層電容特性,但其貢獻小于贗電容特性。當氮磷共摻雜多孔炭膜與聚苯胺復合后,所得復合材料(曲線3與4)的比電容值均有所提高。這是由于氮磷共摻雜多孔炭纖維不僅可以作為高導電率的穩定骨架,也利于聚苯胺的有序分散,而使得復合材料的雙電層電容及贗電容性能都可充分地顯示出來。但具有共價鍵鏈接的復合試樣(曲線4)具有明顯更高的比電容值,這主要歸功于界面共價鍵的存在,將具有骨架穩定性的炭纖維與具有較高贗電容的導電pani材料有效而牢固地結合,兩種材料之間的相互作用增強,會形成更大的π電子離域體系,使兩相間的電子傳遞也由鏈間跳躍模式轉變為鏈內的直接傳導,進而顯著提高電極材料的性能。
圖5及圖6分別為實施例1制得的有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料循環伏安曲線圖和恒流充放電曲線圖。由圖5可看出有共價鍵鏈接的npcm@pani試樣的循環伏安曲線分別在0.1v、0.35v與0.6v有三個還原峰,而在0.15v、0.3v及0.4v有三個氧化峰,這對應于pani的全氧化態/中間摻雜態,中間摻雜態/還原態的氧化還原特征峰。隨著掃描速率的增加,同一電勢下響應電流也成比例增大。圖6是計時電位法測得用具有共價鍵鏈接的npcm@pani雜化材料制備的電極在1a/g、2a/g、5a/g恒定電流下的充放電曲線圖。可看出,具有共價鍵鏈接的npcm@pani雜化材料的充放電曲線均呈現近似于等腰三角形,說明電極反應具有較好的可逆性,而電極電位隨時間呈線性變化,說明該復合材料具有良好的電化學性能。所以本發明的具有共價鍵鏈接的npcm@pani雜化材料可以作為超級電容器的優良電極材料使用。
圖7為實施例1制得的有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料及無共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺復合材料在不同電流密度下的比電容值。明顯可見具有共價鍵鏈接的npcm@pani雜化材料儲能密度遠優于無共價鍵鏈接的npcm/pani復合材料。
實施例2
(1)稱量聚丙烯腈:三苯基膦:sba-15的質量比依次為2.5克:1克:1克,以22克n,n-二甲基甲酰胺為溶劑,在70℃油浴下,溶于n,n-二甲基甲酰胺(dmf)中,用磁力攪拌器以300r/min連續攪拌8h,使混合體系充分溶解并混合均勻,然后用200w的超聲波超聲0.5h,得到聚丙烯腈含量為8%的混合紡絲液。通過靜電紡絲(轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,20號針頭,針頭尺寸為φ0.06mm,電壓25kv,轉速750r/min)獲得納米纖維膜。再通過預氧化、炭化、活化及刻蝕(20%的氫氟酸浸泡24h),等獲得氮磷共摻雜多孔炭膜。
預氧化:在空氣氣氛下,以1℃/min升至280℃恒溫2h;
炭化:在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h;
co2活化:炭化結束后將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降至室溫。
刻蝕:利用20%的氫氟酸將上述活化炭材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,在干燥箱內110℃下干燥過夜得氮磷共摻雜多孔炭膜npcm。
(2)稱取硝酸并配制成16%硝酸水溶液100ml,將其和0.6克氮磷共摻雜活性炭膜混合加入到三口燒瓶中,50℃水浴下恒溫磁力攪拌回流5小時。待反應完全后趁熱過濾,并用蒸餾水洗滌至中性,濾出物在110℃烘箱中干燥12h,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm。
(3)取0.4克上述所得hno3-npcm分散在100ml甲苯與6ml苯胺基甲基三乙氧基硅烷即nd42的混合液中,用玻璃棒將炭膜壓入液面以下并200w超聲分散0.5h;然后加熱至110℃下回流反應24h,50r/min磁力攪拌;之后冷卻、抽濾,用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色;將產物在50℃的真空烘箱中干燥24h,冷卻得到黑色nd42功能化多孔炭膜,記作nd42-npcm。
(4)首先將正己烷與正己醇按5:1(體積比)配成混合溶劑(正己烷/正己醇);將1.98g十六烷基三甲基溴化銨和3.3g樟腦磺酸混合溶于5ml去離子水中,然后加入到30ml的混合溶劑(正己烷/正己醇)中,將0.3g的nd42-npcm和1.2ml苯胺分散至上述的混合體系中,同時在室溫下攪拌4個小時,標記為a溶液。通過相似的過程,b溶液是由1.98g十六烷基三甲基溴化銨和1.5g過硫酸銨溶于5ml去離子水中,并添加30ml的混合溶劑(正己烷/正己醇)中而配制成的。隨后,兩個系統都在冰水浴中攪拌預冷,然后將b溶液以1滴/秒的速度加入到a溶液中。以為300r/min磁力攪拌5分鐘后,整個混合物在大約0~5℃,靜態條件下聚合24小時。過濾沉淀物,緊接著分別用去離子水、乙醇和丙酮進行清洗,最后在60℃下真空干燥24個小時,得到乳液法制備的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
(5)為了檢測制備所得具有共價鍵鏈接的npcm@pani雜化材料電化學性能,將制備得到的npcm@pani作電極材料,以三電極體系進行測試。測試用的電解液為1mol/l的h2so4溶液。
實施例3
(1)稱量聚丙烯腈:三苯基膦:sba-15的質量比依次為2克:1克:1克,以21克n,n-二甲基甲酰胺為溶劑,在70℃油浴下,溶于n,n-二甲基甲酰胺(dmf)中,用磁力攪拌器以300r/min連續攪拌8h,使混合體系充分溶解并混合均勻,然后用200w的超聲波超聲0.5h,得到聚丙烯腈含量為8%的混合紡絲液。通過靜電紡絲(轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,20號針頭,針頭尺寸為φ0.06mm,電壓25kv,轉速750r/min)獲得納米纖維膜。再通過預氧化、炭化、活化及刻蝕(20%的氫氟酸浸泡24h),等獲得氮磷共摻雜多孔炭膜。
預氧化:在空氣氣氛下,以1℃/min升至280℃恒溫2h;
炭化:在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h;
co2活化:炭化結束后將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降至室溫。
刻蝕:利用20%的氫氟酸將上述活化炭材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,在干燥箱內110℃下干燥過夜得氮磷共摻雜多孔炭膜npcm。
(2)稱取硝酸并配制成16%硝酸水溶液100ml,將其和0.5克氮磷共摻雜活性炭膜混合加入到三口燒瓶中,50℃水浴下恒溫磁力攪拌回流5小時。待反應完全后趁熱過濾,并用蒸餾水洗滌至中性,濾出物在110℃烘箱中干燥12h,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm。
(3)取0.35克上述所得hno3-npcm分散在90ml甲苯與4ml苯胺基甲基三乙氧基硅烷即nd42的混合液中,用玻璃棒將炭膜壓入液面以下并200w超聲分散0.5h;然后加熱至110℃下回流反應24h,50r/min磁力攪拌;之后冷卻、抽濾,用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色;將產物在50℃的真空烘箱中干燥24h,冷卻得到黑色nd42功能化多孔炭膜,記作nd42-npcm。
(4)首先將正己烷與正己醇按5:1(體積比)配成混合溶劑(正己烷/正己醇);將1.98g十六烷基三甲基溴化銨和3.3g樟腦磺酸混合溶于5ml去離子水中,然后加入到30ml的混合溶劑(正己烷/正己醇)中,將0.3g的nd42-npcm和1.2ml苯胺分散至上述的混合體系中,同時在室溫下攪拌4個小時,標記為a溶液。通過相似的過程,b溶液是由1.98g十六烷基三甲基溴化銨和1.5g過硫酸銨溶于5ml去離子水中,并添加30ml的混合溶劑(正己烷/正己醇)中而配制成的。隨后,兩個系統都在冰水浴中攪拌預冷,然后將b溶液以2滴/秒的速度加入到a溶液中。以為300r/min磁力攪拌5分鐘后,整個混合物在大約0~5℃,靜態條件下聚合24小時。過濾沉淀物,緊接著分別用去離子水、乙醇和丙酮進行清洗,最后在60℃下真空干燥24個小時,得到乳液法制備的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
(5)為了檢測制備所得具有共價鍵鏈接的npcm@pani雜化材料電化學性能,將制備得到的npcm@pani作電極材料,以三電極體系進行測試。測試用的電解液為1mol/l的h2so4溶液。
實施例4
(1)稱量聚丙烯腈:三苯基膦:sba-15的質量比依次為2克:1克:1克,以21克n,n-二甲基甲酰胺為溶劑,在70℃油浴下,溶于n,n-二甲基甲酰胺(dmf)中,用磁力攪拌器以300r/min連續攪拌8h,使混合體系充分溶解并混合均勻,然后用200w的超聲波超聲0.5h,得到聚丙烯腈含量為8%的混合紡絲液。通過靜電紡絲(轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,20號針頭,針頭尺寸為φ0.06mm,電壓25kv,轉速750r/min)獲得納米纖維膜。再通過預氧化、炭化、活化及刻蝕(20%的氫氟酸浸泡24h),等獲得氮磷共摻雜多孔炭膜。
預氧化:在空氣氣氛下,以1℃/min升至280℃恒溫2h;
炭化:在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h;
co2活化:炭化結束后將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降至室溫。
刻蝕:利用20%的氫氟酸將上述活化炭材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,在干燥箱內110℃下干燥過夜得氮磷共摻雜多孔炭膜npcm。
(2)稱取硝酸并配制成16%硝酸水溶液100ml,將其和0.8克氮磷共摻雜活性炭膜混合加入到三口燒瓶中,50℃水浴下恒溫磁力攪拌回流5小時。待反應完全后趁熱過濾,并用蒸餾水洗滌至中性,濾出物在110℃烘箱中干燥12h,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm。
(3)取0.7克上述所得hno3-npcm分散在100ml甲苯與7ml苯胺基甲基三乙氧基硅烷即nd42的混合液中,用玻璃棒將炭膜壓入液面以下并200w超聲分散0.5h;然后加熱至110℃下回流反應24h,50r/min磁力攪拌;之后冷卻、抽濾,用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色;將產物在50℃的真空烘箱中干燥24h,冷卻得到黑色nd42功能化多孔炭膜,記作nd42-npcm。
(4)首先將正己烷與正己醇按5:1(體積比)配成混合溶劑(正己烷/正己醇);將2g十六烷基三甲基溴化銨和3g樟腦磺酸混合溶于5ml去離子水中,然后加入到30ml的混合溶劑(正己烷/正己醇)中,將0.6g的nd42-npcm和1.2ml苯胺分散至上述的混合體系中,同時在室溫下攪拌4個小時,標記為a溶液。通過相似的過程,b溶液是由2g十六烷基三甲基溴化銨和1.5g過硫酸銨溶于5ml去離子水中,并添加30ml的混合溶劑(正己烷/正己醇)中而配制成的。隨后,兩個系統都在冰水浴中攪拌預冷,然后將b溶液以2滴/秒的速度加入到a溶液中。以為300r/min磁力攪拌5分鐘后,整個混合物在大約0~5℃,靜態條件下聚合24小時。過濾沉淀物,緊接著分別用去離子水、乙醇和丙酮進行清洗,最后在60℃下真空干燥24個小時,得到乳液法制備的氮磷共摻雜多孔炭膜@聚苯胺雜化材料。
(5)為了檢測制備所得具有共價鍵鏈接的npcm@pani雜化材料電化學性能,將制備得到的npcm@pani作電極材料,以三電極體系進行測試。測試用的電解液為1mol/l的h2so4溶液。
實施例5
(1)將聚丙烯腈、三苯基膦和分子篩sba-15在70℃油浴下,溶于n,n-二甲基甲酰胺中,混合均勻,得到復合紡絲液;通過靜電紡絲獲得納米纖維膜,再于空氣氣氛下,以1℃/min自室溫升至280℃恒溫2h;然后在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h。再將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降溫至室溫,最后利用質量濃度20%的氫氟酸將co2活化的材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,干燥,得氮磷共摻雜多孔炭膜。
其中,聚丙烯腈、三苯基膦、sba-15的比為2g:2g:1g,復合紡絲液中聚丙烯腈質量含量為7%;靜電紡絲的條件為:轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,采用20號針頭,針頭尺寸為φ0.06mm,電壓為25kv,轉速為750r/min。
(2)將質量濃度為15%硝酸水溶液和氮磷共摻雜活性炭膜混合后加入到容器中,攪拌回流5小時后趁熱過濾,并用蒸餾水洗滌至中性,干燥,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm;
其中,硝酸水溶液中硝酸與氮磷共摻雜活性炭膜的質量比為15g:0.5g;
(3)將上述所得hno3-npcm分散在甲苯與苯胺基甲基三乙氧基硅烷的混合液中,超聲分散,然后回流反應24h;之后冷卻、抽濾,采用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色,干燥,冷卻得到黑色nd42功能化多孔炭膜;其中,hno3-npcm:甲苯:苯胺基甲基三乙氧基硅烷的比為0.3g:80ml:8ml。
(4)將十六烷基三甲基溴化銨和樟腦磺酸加入到水中混合,然后加入到正己烷和正己醇的混合液中(正己烷和正己醇的體積比為5:1),得到混合物;將苯胺基甲基三乙氧基硅烷功能化多孔炭膜和苯胺分散至上述的混合物中,在室溫下200r/min下攪拌4h,形成a溶液;其中,十六烷基三甲基溴化銨、樟腦磺酸、水、正己烷和正己醇的混合液、苯胺基甲基三乙氧基硅烷功能化多孔炭膜、苯胺的比為2g:3g:4ml:25ml:0.25g:2ml;
(5)將十六烷基三甲基溴化銨和過硫酸銨加入到水中,然后添加至正己烷和正己醇的混合液中(正己烷和正己醇的體積比為5:1),形成b溶液;其中,十六烷基三甲基溴化銨、過硫酸銨、水、正己烷和正己醇的混合液的比為1.8g:1.7g:4ml:30ml;
(6)將a溶液和b溶液在冰水浴中攪拌預冷至0℃后,將b溶液以1滴/秒的速度滴加到a溶液中,攪拌5分鐘后,在0℃,靜態條件下聚合25h后過濾沉淀物,分別用去離子水、乙醇和丙酮進行清洗,最后干燥,得到氮磷共摻雜多孔炭膜@聚苯胺雜化材料;其中,樟腦磺酸與過硫酸銨的比為3g:1.7g。
實施例6
(1)將聚丙烯腈、三苯基膦和分子篩sba-15在70℃油浴下,溶于n,n-二甲基甲酰胺中,混合均勻,得到復合紡絲液;通過靜電紡絲獲得納米纖維膜,再于空氣氣氛下,以1℃/min自室溫升至280℃恒溫2h;然后在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h。再將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降溫至室溫,最后利用質量濃度20%的氫氟酸將co2活化的材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,干燥,得氮磷共摻雜多孔炭膜。
其中,聚丙烯腈、三苯基膦、sba-15的比為3g:1.5g:2g,復合紡絲液中聚丙烯腈質量含量為12%;靜電紡絲的條件為:轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,采用20號針頭,針頭尺寸為φ0.06mm,電壓為25kv,轉速為750r/min。
(2)將質量濃度為18%硝酸水溶液和氮磷共摻雜活性炭膜混合后加入到容器中,攪拌回流5小時后趁熱過濾,并用蒸餾水洗滌至中性,干燥,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm;
其中,硝酸水溶液中硝酸與氮磷共摻雜活性炭膜的質量比為20g:0.7g;
(3)將上述所得hno3-npcm分散在甲苯與苯胺基甲基三乙氧基硅烷的混合液中,超聲分散,然后回流反應24h;之后冷卻、抽濾,采用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色,干燥,冷卻得到黑色nd42功能化多孔炭膜;其中,hno3-npcm:甲苯:苯胺基甲基三乙氧基硅烷的比為0.8g:120ml:5ml。
(4)將十六烷基三甲基溴化銨和樟腦磺酸加入到水中混合,然后加入到正己烷和正己醇的混合液中(正己烷和正己醇的體積比為5:1),得到混合物;將苯胺基甲基三乙氧基硅烷功能化多孔炭膜和苯胺分散至上述的混合物中,在室溫下300r/min下攪拌4h,形成a溶液;其中,十六烷基三甲基溴化銨、樟腦磺酸、水、正己烷和正己醇的混合液、苯胺基甲基三乙氧基硅烷功能化多孔炭膜、苯胺的比為2.2g:3.5g:5ml:30ml:0.4g:1ml;
(5)將十六烷基三甲基溴化銨和過硫酸銨加入到水中,然后添加至正己烷和正己醇的混合液中(正己烷和正己醇的體積比為5:1),形成b溶液;其中,十六烷基三甲基溴化銨、過硫酸銨、水、正己烷和正己醇的混合液的比為2g:1.5g:5ml:25ml;
(6)將a溶液和b溶液在冰水浴中攪拌預冷至5℃后,將b溶液以2滴/秒的速度滴加到a溶液中,攪拌5分鐘后,在5℃,靜態條件下聚合23h后過濾沉淀物,分別用去離子水、乙醇和丙酮進行清洗,最后干燥,得到氮磷共摻雜多孔炭膜@聚苯胺雜化材料;其中,樟腦磺酸與過硫酸銨的比為3.5g:1.5g。
實施例7
(1)將聚丙烯腈、三苯基膦和分子篩sba-15在70℃油浴下,溶于n,n-二甲基甲酰胺中,混合均勻,得到復合紡絲液;通過靜電紡絲獲得納米纖維膜,再于空氣氣氛下,以1℃/min自室溫升至280℃恒溫2h;然后在氬氣氣氛條件下,以5℃/min從280℃升至850℃,恒溫0.5h。再將氬氣切換成co2氣氛,以5℃/min升至900℃,恒溫30min,再把co2換成氬氣,在氬氣氣氛下降溫至室溫,最后利用質量濃度20%的氫氟酸將co2活化的材料浸泡24h,刻蝕去除介孔模板劑sba-15,然后水洗抽濾至中性,干燥,得氮磷共摻雜多孔炭膜。
其中,聚丙烯腈、三苯基膦、sba-15的比為4g:1g:1.5g,復合紡絲液中聚丙烯腈質量含量為10%;靜電紡絲的條件為:轉鼓軸心距紡絲針頭距離為15cm,轉鼓為φ8.5×16cm,采用20號針頭,針頭尺寸為φ0.06mm,電壓為25kv,轉速為750r/min。
(2)將質量濃度為20%硝酸水溶液和氮磷共摻雜活性炭膜混合后加入到容器中,攪拌回流5小時后趁熱過濾,并用蒸餾水洗滌至中性,干燥,冷卻后得到黑色硝酸活化多孔炭膜,取出待用,并記作hno3-npcm;
其中,硝酸水溶液中硝酸與氮磷共摻雜活性炭膜的質量比為17g:1g;
(3)將上述所得hno3-npcm分散在甲苯與苯胺基甲基三乙氧基硅烷的混合液中,超聲分散,然后回流反應24h;之后冷卻、抽濾,采用甲苯、丙酮、蒸餾水依次洗滌,直至濾液無色,干燥,冷卻得到黑色nd42功能化多孔炭膜;其中,hno3-npcm:甲苯:苯胺基甲基三乙氧基硅烷的比為0.5g:100ml:3ml。
(4)將十六烷基三甲基溴化銨和樟腦磺酸加入到水中混合,然后加入到正己烷和正己醇的混合液中(正己烷和正己醇的體積比為5:1),得到混合物;將苯胺基甲基三乙氧基硅烷功能化多孔炭膜和苯胺分散至上述的混合物中,在室溫下400r/min下攪拌4h,形成a溶液;其中,十六烷基三甲基溴化銨、樟腦磺酸、水、正己烷和正己醇的混合液、苯胺基甲基三乙氧基硅烷功能化多孔炭膜、苯胺的比為1.8g:3.2g:6ml:35ml:0.5g:1.5ml;
(5)將十六烷基三甲基溴化銨和過硫酸銨加入到水中,然后添加至正己烷和正己醇的混合液中(正己烷和正己醇的體積比為5:1),形成b溶液;其中,十六烷基三甲基溴化銨、過硫酸銨、水、正己烷和正己醇的混合液的比為2.2g:1.3g:6ml:35ml;
(6)將a溶液和b溶液在冰水浴中攪拌預冷至3℃后,將b溶液以1滴/秒的速度滴加到a溶液中,攪拌5分鐘后,在3℃,靜態條件下聚合24h后過濾沉淀物,分別用去離子水、乙醇和丙酮進行清洗,最后干燥,得到氮磷共摻雜多孔炭膜@聚苯胺雜化材料;其中,樟腦磺酸與過硫酸銨的比為3.2g:1.3g。
所制得的具有界面共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料,其特征在于,在氮磷共摻雜多孔炭膜與聚苯胺兩相界面之間通過共價鍵結合,且雜化材料中聚苯胺以微纖均勻包覆在炭纖維膜表面。
本發明的原理是:有共價鍵鏈接的氮磷共摻雜多孔炭膜@聚苯胺雜化材料合成原理如下圖8所示。多孔炭纖維膜經過硝酸溶液處理后,炭纖維的表面含有豐富的羧基和羥基,可以與nd42水解后的羥基縮合,使偶聯劑以化學鍵接枝在炭材料的表面。另外,nd42分子上的活性苯胺基團又可以和苯胺單體進行共聚反應,使樟腦磺酸摻雜的pani原位接枝在多孔炭纖維膜的表面,形成具有共價鍵鏈接的牢固均勻包覆層。由于有界面共價鍵的存在,將具有骨架穩定性的多孔炭纖維與具有較高贗電容的導電pani材料有效而牢固地結合,兩種材料之間的相互作用增強,會形成更大的π電子離域體系,使兩相間的電子傳遞也由鏈間跳躍模式轉變為鏈內的直接傳導,進而大幅度提高電極材料的比電容及循環穩定性。這對于電池材料、光電轉換材料性能的提升均非常有利。
盡管本發明的實施方案已公開如上,但其并不僅僅限于說明書和實施方式中所列運用,它完全可以被適用于各種適合本發明的領域,對于熟悉本領域的人員而言,可容易地實現另外的修改,因此在不背離權利要求及等同范圍所限定的一般概念下,本發明并不限于特定的細節和這里示出與描述的圖例。
- 還沒有人留言評論。精彩留言會獲得點贊!