基于人工智能片段化技術的先導活性分子生成與篩選方法
本發明屬于ai輔助藥物設計領域,特別是涉及基于人工智能片段化技術的活性分子生成與篩選方法,包括基于aidd的片段化活性分子的數據處理、高親和力的活性分子片段預測以及先導活性分子篩選。
背景技術:
1、cadd(computer?aided?drug?design):計算機輔助藥物設計,依據生物化學、酶學、分子生物學以及遺傳學等生命科學的研究成果,針對這些基礎研究中所揭示的包括酶、受體、離子通道及核酸等潛在的藥物設計靶點,并參考其它類源性配體或天然產物的化學結構特征,以計算機化學為基礎,通過計算機的模擬、計算和預算藥物與受體生物大分子之間的相互作用,考察藥物與靶點的結構互補、性質互補等,設計出合理的藥物分子。
2、隨著人工智能(ai)的蓬勃發展,aidd(aidrug?discovery&design)已經介入到新藥設計到研發的大部分環節當中,為新藥發現與開發帶來了極大的助力。在各種人工智能方法中,生成模型(generation?model)近年來備受關注。在這些成功的啟發下,研究者們現在將生成模型技術應用于藥物從頭設計,這被認為是藥物發現的起源。在這個視角中,如遞歸神經網絡、自編碼器、生成對抗網絡、transformer和帶有強化學習的混合模型在各個分子生成任務中都發揮了非常出色的功能。
3、分子表示是分子生成工作流程中的關鍵任務。研究人員通過從1d,2d,3d和圖像等多個維度和視角出發構建了分子精確表征的模型,并通過實驗進行了驗證。但是大多數人關注的是以原子和鍵為基礎結構的分子表征方法,而忽略了分子結構片段的相互作用對分子性質的影響。
4、在分子表示的演變歷程中,最初使用通用名稱對分子進行命名,但隨著化學領域的發展,1919年international?union?of?pure?and?applied?chemistry(iupac)的成立標志著對化學命名法和術語的規范化。然而,這種命名法繁瑣難懂,推動了更適合計算機的數字表示的發展,其中包括基于分子圖的smiles(simplified?molecular?input?line?entryspecification)格式和3d分子格式。smiles通過字符串簡潔而可讀地表示分子,但同一分子可有多個等效的smiles表示,引發了其他格式的發展,如smiles?arbitrary?targetspecification(smarts)、smiles?inreaction?kekule?system(smirks)、self-referencing?embedded?strings(selfie)s等。此外,iupac創建的internationalchemical?identifier(inchi)格式提供了機器可讀的分子表示,包含電荷、立體化學和同位素信息。另一方面,3d分子格式如mol和mol2包含了原子在3d空間中的位置,適用于包含更多信息的領域,如晶體學和分子動力學。目前,smiles和圖形編碼已成為神經網絡模型中常用的小分子表示方法。
5、基于1d序列的分子編碼方法是將藥物表示問題描述為序列編碼問題。這類方法中的大多數都是基于簡化的分子輸入行輸入系統(smiles),這是一種使用短的americanstandard?code?for?information?interchange(ascii)字符串weininger描述分子結構的行符號。字符變化自動編碼器(c-vae)逐個字符地生成smiles字符串;grammar?vae(g-vae)中kusner等人遵循上下文無關文法給出的句法限制生成smiles字符串;句法指導的vae(sd-vae)中,dai等人將smiles的句法和語義通過屬性語法約束結合在一起。然而,由于基于序列的方法大多數是基于smiles字符串進行編碼,而smiles字符串是一種具有嚴格語法規則的編碼方式,因此面臨兩個重要問題:一是有效性問題,生成模型會錯誤地理解smlies序列語義,生成錯誤的smiles編碼。二是smiles的非結構化性質使得兩個相似的分子極有可能完全不同。
6、分子由片段構成,片段是分子的展現出各種性質的基本化學結構。針對片段的分子研究,可以掌握分子內部或分子間局部片段之間的相互作用。分子間產生的相互作用來源于分子片段間的相互作用的統一,因此,高親和度的片段是分子親和力的重要源頭。構建一個基于高親和力分子片段生成的分子,將會是潛在的高親和力的分子。
7、強化學習是一種以環境反饋為輸入,自適應環境的面向目標的機器學習方法。分子生成的結構約束通過強化學習反復獎勵更加接近需要約束結構的生成分子來完成。
8、為了在生成的分子中保留高親和度分子片段的結構特征,通過對生成分子和目標片段谷本相似度的約束進行強化學習。模型每一次生成的新分子在結構上越接近目標片段,則agent進行積極獎勵,而生成的新分子遠離目標片段,則進行懲罰。使用強化學習避免了直接連接目標片段的低自由度方法,從而避免模型生成全新分子的能力被限制情況。一個結構受限的分子生成策略是限制輸出分子包含一個特定的骨架或片段。langevin等人和li等人建立了生成模型,輸出具有特定骨架的藥物分子。這些骨架通常是從具有良好生物特性的現有藥物中提取的。jin、podda、imrie和green等人也開發了基于骨架的生成模型,學習生成具有特定片段的分子。但是,基于結構約束的分子生成模型往往會產生大量重復的結構和分子,因其限定了分子骨架的主要結構,會約束生成模型的自由度,導致生成同一藥物分子的大量同類分子,從而降低模型對新藥物的學習和生成能力。因此,在生成模型中考慮多種可能的高親和力片段并以此為基礎生成分子,通過強化學習的應用使生成的分子既能保留目標片段的結構特征,又能基于固定結構特征產生變化,提高模型生成分子的自由度。
9、本技術發明人的前期研究公開了一種融合復合蛋白質相互作用的親和力預測模型fotf-cpi,基于最佳運輸的片段化方法來提高模型對化合物和蛋白質序列的理解(fotf-cpi:a?compound-protein?interaction?prediction?transformer?based?on?the?fusionof?optimal?transport?fragments,iscience,volume?27,issue?1,19january?2024)。該模型采用bpe方法,存在詞粒度錯位、詞表構建效率低、低泛化和無效片段多的問題。
10、此外,以protac藥物設計模型為例,目前常見的藥物預測模型通常存在如下缺陷:
11、1.傳統protac設計通常需要考慮多種復雜因素,如目標蛋白和e3連接酶之間的相互作用、連接子長度和柔性等。這使得設計過程繁瑣耗時。
12、2.傳統protac設計倚賴反復合成和測試多個結構變體,甚至通過濕實驗去驗證以找到最優組合,這種方法效率低、優化周期長。
13、3.傳統protac設計往往面臨不可預測的挑戰,如連接子對靶標蛋白與e3連接酶之間的相互作用的影響,這讓設計過程具有較大的不確定性和風險。
技術實現思路
1、本發明針對現有技術不足,從語言模型角度,將分子碎片化為多個token的組合,關注這些token片段的相互作用關系,找到對分子性質影響較大的片段。進一步以分子的分段表示為基礎結合語言模型,使用基于語言模型的親和力模型來篩選具有高親和力的分子片段。所得到高親和力的分子片段可以進一步結合分子屬性約束,最終,通過多維屬性預測模型對初步生成的分子進行篩選,生成能夠降解與疾病相關的靶蛋白的protac分子,從而實現了ai輔助protac藥物生成的完整解決方案。
2、為了實現本發明目的,本發明首先提供了基于aidd的片段化活性分子數據處理方法,包括如下步驟:
3、步驟一、構建片段詞典
4、(1)將數據集中的數據源分子轉化為線性化文本,數據源分子為化合物、核酸、多糖中的一種或幾種的組合時,將數據源分子轉化為smiles字符串,數據源分子為多肽或蛋白質時,將多肽或蛋白質中氨基酸殘基序列按照亞基順序首尾相接,形成完整的一條氨基酸殘基序列;(2)采用序列切分算法將所有smiles字符串或氨基酸殘基序列切分為片段,構建片段詞典,訓練過程如下:
5、a)根據預生成token的頻率對所有候選token從高到低排名;
6、b)所有具有概率的token都用于在最優傳輸算法中初始化,在每個時間步上,根據傳輸矩陣得到熵最大的片段詞典;
7、c)窮舉所有的時間步長,選擇滿足指定指數搜索空間的詞匯作為最終詞匯;
8、d)使用貪婪策略編碼文本:先將句子分割成字符級的標記,若合并后的token在片段詞典中,則將兩個連續的token合并為一個token,直到沒有token可以合并為止,片段詞典外標記將被分割成更小的標記。例如,如果token“cc(=o)nc”和“cc1=cn”是兩個相鄰的token,且兩者都在片段詞典中,那么兩者前后相接組成的序列將組合為新的token,即“cc(=o)nc?cc1=cn”;
9、步驟二、活性分子數據處理
10、(1)活性分子轉化為線性化文本,活性分子為化合物、核酸、多糖中的一種或幾種的組合時,將活性分子轉化為smiles字符串,活性分子為多肽或蛋白質時,將多肽或蛋白質中氨基酸殘基序列按照亞基順序首尾相接,形成完整的一條氨基酸殘基序列;
11、(2)在步驟一構建的片段詞典指導下,采用序列切分算法將活性分子的smiles字符串或氨基酸殘基序列切分為片段,統計詞頻低于5個的片段定義為“低頻片段”,在編碼過程中,包含“低頻片段”的活性分子中所有“低頻片段”按照同一個token進行編碼,得到活性分子片段;(3)經過分子片段表征提取方法提取活性分子每個片段的表征。優選分子片段表征提取方法選自word2vec、ernie、electra、universal?sentence?encoder、bag?of?words、tf-idf、n-gram、glove、bert、gpt、elmo或transformer。本發明一個具體的示例,分子片段表征提取方法為transformer。
12、上述方法,數據集可以為protein?data?bank(pdb)、uniprot、pfam、interpro、ncbi?protein、swiss-model?repository、kegg、string、bindingdb、pdbbind、dud-e、zinc、pubchem、chembl、drugbank、chemspider、hmdb(human?metabolome?database)、pubchem、bioassay、molport、genbank、ensembl、ucsc?genome?browser、ddbj(dna?data?bank?ofjapan)、1000genomes?project、gencode、human?genome?project(hgp)、gtex(genotype-tissue?expression)、tcga(the?cancer?genome?atlas)、dbsnp、arrayexpress、iedb(immune?epitope?database)、vaxign、antigendb、protegen、cancer?antigenic?peptidedatabase(caped)、epimhc、viruses.hostdb、allergenonline、mhcbn、hla?ligand?atlas、antibodypedia、abcam、the?antibody?registry、imgt/2d-structure-db、cab(chineseantibody?database)、sabdab(structural?antibody?database)、cov-abdab、adrecs-target、abminer、iuphar/bps?guide?to?pharmacology、therapeutic?antibody?database(tadb)、oas(observed?antibody?space)。
13、本發明一個具體的示例,先導活性分子為小分子化合物和蛋白質,小分子化合物數據集為zinc?clean?lead,蛋白質數據集為pdb。
14、優選對數據集中的小分子化合物進行篩選,例如1)除去碳、氮、硫、氧、氟、溴和氫以外,含有帶其他電原子的分子被剔除;2)分子量在200~600之間的類藥化合物;3)logp(通過rdkit計算)在-2~6之間等。
15、優選所述smiles字符串為canonical?smiles格式。
16、本發明所述方法將數據源分子或先導活性分子轉化為smiles字符串時,還包括對smiles字符串進行解析和切割:對先導活性分子主鏈中包含支鏈的位置填入字符“r”保留主鏈中的支鏈信息,支鏈中與主鏈相接的位置保留“(”用于保存支鏈中的拓撲信息,按照主鏈、支鏈的順序將切割后的smiles進行首尾相接,形成一條smiles字符串。
17、本發明所述的方法,所述序列切分算法可以為volt、bpe、character?slicing(cs)、recap、brics(bcs)、根據院子間反應分數切分、emolfrag、fcs、bpe_nlm、macfrag、fgsplitting、spe、tree?decomposition。本發明一個具體的示例為volt。
18、本發明所述的方法,構建片段詞典訓練過程中優選采用byte?pair?encoder(例如bpe-100k)、bpe_nlm、fcs生成的token作為候選。
19、本發明所述的方法,構建片段詞典訓練過程中考慮到最優傳輸算法中包含的松弛策略,會導致不符合規范的傳輸情況。優選刪除分布字符小于0.001頻率的token。
20、本發明另一目的在于提供上述方法處理得到的先導活性分子片段數據。
21、本發明另一目的在于提供所述基于aidd的先導活性分子片段數據在藥物設計中的應用。
22、基于上述方法獲得的先導活性分子片段可進一步用于優化分子結構、預測靶標相互作用、設計組合新的功能性分子片段、揭示分子機制和作用方式、生物標志物研究、分子動力學模擬、設計開發新型材料、研究片段降解路徑和行為。
23、本發明另一目的在于提供對上述先導活性分子片段進行親和力預測的方法,將先導活性分子片段的表征輸入蛋白-化合物親和力模型提取相應信息,選擇a、b兩種先導活性分子的片段集合代表a、b兩組片段,組間兩兩配對(例如小分子化合物和蛋白質),或同一先導活性分子片段分為a、b兩組,組間兩兩配對,根據配對片段之間相互作用進行計算,所述蛋白-化合物親和力模型使用基于預測結果和真實標簽的二元交叉熵損失函數對整個網絡進行持續優化,最終獲得各配對片段的親和度特征。
24、上述蛋白-化合物親和力模型可以為線性回歸(linear?regression)、邏輯回歸(logistic?regression)、決策樹(decision?trees)、隨機森林(random?forests)、梯度提升機(gradient?boosting?machines,gbm)、支持向量機(support?vector?machines,svm)、k近鄰算法(k-nearest?neighbors,k-nn)、樸素貝葉斯(bayes)、全鏈接神經網絡(fully?connected?neural?networks,fcnn)、卷積神經網絡(convolutional?neuralnetworks,cnns)、循環神經網絡(recurrent?neural?networks,rnns)、長短期記憶網絡(long?short-term?memory,lstm)、圖神經網絡(graph?neural?networks,gnns)、變分自編碼器(variational?autoencoders,vaes)、生成對抗網絡(generative?adversarialnetworks,gans)、transformer模型、l2、deepdta、widedta、deepaffinity、graphdta、gat-dta、moltrans、simboost、matchmaker、deepconv-dti、transformercpi、deepcpi、graphbar、molaical、attentiondta、kg-dti、drugvqa、fotf-cpi、monn、dl-cpi、dimenet、geometric?gnn、metadti、deeppurpose、mt-dti、bionev、cpi-gnn、gcn、iifdti。
25、本發明一個具體的示例,以蛋白-化合物親和力模型fotf-cpi為基礎,結合本發明所述的活性分子數據預處理方法,構建基于volt算法的fotf-cpi,即fotf-cpi(volt)。
26、所述蛋白-化合物親和力模型預先使用蛋白-化合物庫進行訓練。所述蛋白-化合物庫可以為casf,bindingdb,chembl,pubchem,dud-e,pdbbind,kiba或davis。本發明一個具體的示例,所述蛋白-化合物親和力庫為bindingdb。
27、優選二元交叉熵損失函數公式如下:
28、
29、其中yi是預測的結果值,li是真實的標簽值。
30、上述親和力預測的方法,具體包括如下步驟:
31、(1)分別得到a組片段表征的全局注意力矩陣和b組片段表征的全局注意力矩陣;
32、(2)將a組片段表征的全局注意力矩陣和b組片段表征的全局注意力矩陣相乘得到局部親和度矩陣(為了避免局部親和度矩陣的親和度得分過高,優選采取歸一化處理);
33、(3)將局部親和度矩陣通過softmax和轉置得到局部注意力修正矩陣;
34、(4)將局部注意力修正矩陣與a組片段表征的全局注意力矩陣相乘得到局部注意力修正下的a組片段表征矩陣;將局部注意力修正矩陣與b組片段表征的全局注意力矩陣相乘得到局部注意力修正下的b組片段表征矩陣;
35、(5)將a組片段表征的全局注意力矩陣與局部注意力修正下的a組片段表征矩陣進行向量維度上的拼接,得到混合a組片段表征;將b組片段表征的全局注意力矩陣與局部注意力修正下的b組片段表征矩陣進行向量維度上的拼接,得到混合b組片段表征;
36、(6)分別將混合a組片段表征和混合b組片段表征通過全連接層后通過全局自適應池化,得到全局特征與局部特征融合后的a組片段表征和b組片段表征;
37、(7)將全局特征與局部特征融合后的a組片段表征和b組片段表征在片段維度上進行拼接,得到每一對a組和b組片段的親和度特征;將得到的親和度特征,依次通過全局自適應池化層與激活函數層得到每一對a組和b組片段的預測結果,依據預測的結果和真實標簽,使用二元交叉熵損失函數,不斷優化整個網絡,最終獲得各配對片段的親和度特征。
38、本發明一個具體的示例,兩種活性分子分別為小分子化合物和蛋白質,具體過程為分別得到分子片段的全局注意力矩陣(即小分子化合物片段表征)c和蛋白質片段的全局注意力矩陣(即蛋白質片段表征)p。然后將小分子化合物片段表征c和蛋白質片段表征p相成得到局部親和度矩陣a。然后將矩陣a通過softmax和轉置等處理得到局部注意力修正矩陣b。將矩陣b與分子片段的全局注意力矩陣c相乘得到局部注意力修正下的小分子化合物片段表征c'。同樣,將b與蛋白片段的全局注意力矩陣p相乘得到局部注意力修正下的蛋白質表征p'。然后將c和c'進行向量維度上的拼接,得到混合(即局部和全局注意力修正的)小分子化合物表征,將p和p'拼接得到混合(即局部和全局注意力修正的)蛋白質表征。拼接得到的混合小分子化合物表征和混合蛋白質表征通過全連接層后通過全局自適應池化,分別得到全局特征與局部特征融合后的小分子化合物表征和蛋白質表征。將融合得到的小分子化合物表征和蛋白質表征在片段維度上進行拼接,得到親和度特征。將得到的親和度特征,依次通過全局自適應池化層與激活函數層得到一對小分子化合物和蛋白質的預測結果,依據預測的結果和真實標簽,結合二元交叉熵損失函數,不斷優化整個網絡。
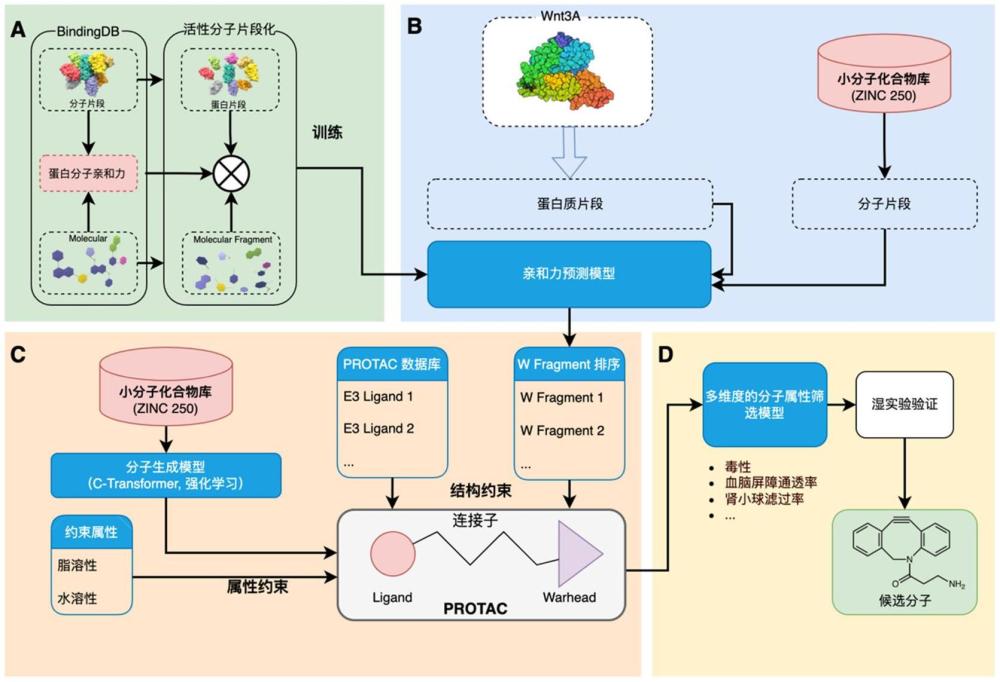
39、本發明所述方法進行高親和力活性分子片段預測示意圖如圖1所示。
40、本發明另一目的在于提供上述方法預測得到的高親和力活性分子片段。
41、本發明另一目的在于提供所述高親和力活性分子片段在藥物設計中的應用。
42、本發明另一目的在于提供一種先導活性分子的預測方法,可基于本發明所述的方法獲得的活性分子片段的表征,或高親和力活性分子片段表征,經分子生成模型生成先導活性分子。
43、所述分子生成模型可以為gru、lstm、variaational?autoencoders(vaes)、generative?adversarial?networks(gans)、junction?tree?variational?autoencoder(jtvae)、smiles-based?models、graphvae、graphgan、reinforcement?learning(rl)models、molecular?transformer、deepchem?models、self-organizing?maps(soms)、latent?space?optimization?models、cvae(conditional?vae)、c-transformer(conditional?transformer)。
44、本發明一個具體的示例,以分子生成c-transformer為基礎,引入活性分子的至少兩個屬性作為c-transformer的條件編碼,將活性分子線性化文本smiles切分為片段表征,作為c-transformer的結構編碼訓練,使用c-transformer對一個隨機初始分子和任意一個片段進行編碼,計算初始分子和片段距離d,計算隨機初始分子分子骨架和片段分子骨架的相似度s,通過強化學習對d*s數值作為獎勵參照,相似度大且距離近則進行獎勵,反之進行懲罰,從而對生成的分子結構進行約束。
45、所述活性分子的屬性選自脂溶性、水溶性、分子量、溶解度、分配系數(logp)、pka值、極性表面積(psa)、氫鍵供體數量、氫鍵受體數量、拓撲極性表面積(tpsa)、半衰期、降解速率常數、熱穩定性、ph穩定性、光穩定性、體外代謝半衰期、內在清除率、生物利用度、毒性、血腦屏障通透性、水溶液酸堿性、溶解度、極性。
46、所述c-transformer中對結構約束的損失函數公式如下:
47、
48、其中d為初始分子和片段的歐式距離,s為分子骨架與片段分子骨架的相似度,a為用于分配c-transformer和強化學習對模型梯度影響的權重參數,n為batch的大小。
49、a可通過以0.1為單位遍歷,通過通過人工對生成模型的骨架和約束片段骨架的相似度評估獲得。
50、進一步的,所述c-transformer中對分子屬性的損失函數公式如下:
51、
52、其中ai是目標屬性值,yi是生成分子計算得出的屬性。
53、總的loss函數表示為如下公式:
54、
55、本發明所述的預測方法還包括對經分子生成模型生成的先導活性分子進一步篩選的步驟。
56、所述的預測方法,可以通過合成活性分子并進行屬性測試的方法篩選,也可以使用篩選模型進行篩選,所述篩選模型選自gcn、mpnn、dmpnn、mdam-sum、mdam-cat、transformer?based?models(molecular?transformer\chemformer)、cmpnn、weave、compt、chemberta、molbert、random?forests、gradient?boosting、xg-boost或mdam。
57、mdam(multi-dimensional?attention?mechanism)是一種基于注意力機制多維特征融合的分子性質預測模型。對先導活性分子的1d,2d和3d信息進行采樣構建多維屬性融合,其中對先導活性分子的3d結構采樣使用spherenet以及注意力機制生成3d結構特征網絡,通過預訓練獲得分子的3d結構的特征向量。(如圖2所示)圖2展示了本發明所述方法使用mdam框架的活性分子設計示意,主要包括五個部分:(a)從化合物中獲取的分子smiles1d編碼序列、2d圖結構和3d圖結構,(b)分子1d序列特征向量生成模塊,(c)分子2d原子圖特征向量生成模塊,(d)分子3d結構特征向量生成模塊,(e)多維特征融合模塊。
58、基于注意力的多維特征編碼器的輸入是分為三個通道的smiles。通道(i):為了處理smiles字符串,mdam使用頻繁連續子序列(fcs)算法將smiles編碼生成序列數據,作為序列編碼器的輸入。序列編碼器使用transformer對序列數據進行編碼,直接獲得分子1d序列的特征向量。通道(ii):rdkit將smiles字符串轉換為分子圖,作為圖形編碼器的輸入。通道(iii):mdam使用spherenet作為3d網絡來生成3d分子結構,處理分子的3d坐標和其他結構信息。從這三個通道獲得的特征向量分別為:1d序列特征向量、2d原子圖特征向量和3d結構特征向量。特征融合模塊用于融合這些向量并預測下游的分子性質。
59、在使用mdam對生成的分子進行篩選的過程中,首先,通過rdkit軟件,將獲得的分子smiles編碼轉換為1d的序列、2d的圖結構和3d的院子坐標結構。分別作為三個通道輸入到訓練好的mdam模型中,由模型輸出對應預測屬性,并根據分子屬性所需閾值對分子進行篩選。
60、本發明另一目的在于提供一種生物活性分子,采用本發明所述的方法預測得到。
61、基于本發明公開的上述多種方法,本發明還提供了一種protac分子設計方法,通過對靶蛋白、e3連接酶以及雙功能小分子片段化數據處理,基于親和力、物化屬性、分子結構以及多維屬性多重約束進行篩選或預測。
62、為了實現上述目的,根據本技術的另一方面,提供了一種活性分子生成裝置,該裝置包括:預測單元,用于預測兩個活性分子之間配對的分子片段或活性分子不同片段配對的分子片段親和力;生成單元:定義目標分子的一個或多個屬性,基于所述一個或多個屬性生成先導分子;篩選單元:定義目標分子的一個或多個屬性,基于所述一個或多個屬性標準篩選出先導活性分子。
63、為了實現上述目的,根據本技術的另一方面,提供了一種處理器,所述處理器用于運行程序,其中,所述程序運行時執行上述的片段親和力預測、分子生成和屬性篩選方法。
64、為了實現上述目的,根據本身去的另一方面,提供了一種計算機可讀存儲介質,其特征在于,所述計算機可讀存儲介質包括存儲的程序,其中,在所述程序運行時控制所述計算機可讀存儲介質所在設備執行上述所述的片段親和力預測、分子生成和屬性篩選方法。
65、本發明一個具體的示例,基于上述方法,得到了一類活性分子,結構如下:
66、
67、本發明優點:
68、本發明使用基于ai的活性分子相互作用預測、活性分子生成與活性分子屬性預測,與目前常用的基于ai的protac生成方法相比,具備以下優勢:
69、1.使用了基于片段的設計方法:在全流程protac設計中,片段的引入使得結構多樣性增加,能夠允許ai模型從更廣泛的化學空間中選擇和優化分子片段,從而生成更加多樣化的protac分子。其通過逐步優化和篩選小分子片段,可以更快速找到最佳的蛋白降解劑的組合,這比傳統方法更具系統性和高效性,方便快速篩選出有效的protac分子。基于片段的方法也能夠進行模塊化設計,允許模型或者研究人員以模塊化的方式進行組合和優化,可以更靈活的設計和合成protac分子。
70、2.使用了基于結構約束的分子生成方法:片段結構源于真實小分子片段,通過對生成的分子施加對這些片段結構的約束能夠提高生成分子的結構合理性。合理切分的片段結構能夠保留功能分子團的重要特性,通過結構約束能夠幫助保留關鍵功能團的位置和幾何形狀,從而確保分子與靶標蛋白的有效結合和相互作用。結構的約束可以顯著減少生成分子的化學空間,從而加速分子的篩選和優化過程。結構約束同時能夠提高分子合成的可行性,合理的結構約束可以避免難以通過現有化學方法合成的分子,從而提高濕實驗驗證的成功率。
71、3.使用了基于屬性約束的分子生成方法:能夠有效提高先導分子的質量,優化藥物性質,通過設置屬性約束,可以確保生成的分子具有理想的物理化學和藥代動力學性質,如良好的溶解度、生物利用度和代謝穩定性。同時,也避免了無效分子的生成,屬性約束可以過濾掉那些不符合關鍵藥物屬性的分子,從而減少不具備開發潛力的的分子的生成,節省資源和時間。通過對合成可行性的約束,能夠有效提高合成分子的成功率,減少試驗成本。通過該方法,減少了人工對生成分子的篩選,減少篩選過程中的主觀偏見,提高藥物設計的客觀性和科學性。
72、4.本發明在片段化活性分子數據處理方法過程中使用volt算法,(1)在對齊字節粒度和詞粒度方面,結合了字節級別和詞級別的對齊信息,有別于現有技術使用bpe基于字節對的合并。通過對齊,volt能夠更好地捕捉詞語的語義和結構信息。(2)volt利用最優傳輸(optimal?transport)理論,通過高效的數學優化方法構建詞匯表,使其在處理大規模語料時表現更優。(3)volt算法通過在詞匯表構建過程中考慮不同語言的多樣性和復雜性,能夠生成更具泛化能力的詞匯表,適用于多語言和跨領域的自然語言處理任務。(4)減少數據碎片化:bpe有時會將一些常見的詞分割成不常見的子詞單元,而volt能夠通過優化避免這種不必要的分割,從而保持詞語的完整性和連貫性。
73、5.本發明采用片段化方式對活性分子進行處理,其優點在于:
74、(1)化合物庫小而多樣,通過片段化技術使用較小的化合物庫,能夠獲取到多樣性的片段,且片段庫更容易管理。
75、(2)更高的命中率,片段較小且簡單,可以更容易適應蛋白質的不同口袋,從而增加了與靶點結合的可能性。
76、(3)優化的起點,片段化處理后識別的命中物,可以作為優化的起點,可以通過化學合成進一步擴展和優化,能夠提高親和力和特異性。
77、(4)便于發現新穎的結合位點。較小的片段結構,能夠識別傳統hts方法可能遺漏的結合位點,可以探索個蛋白質表面更多的區域。
78、(5)結構簡單易于合成,分子片段通常結構簡單,這是的他們的化學合成更加容易,通過合成和測試衍生物,可以快速進行結構優化。
79、6.本發明采用基于片段詞典的序列切分算法,其優點在于:
80、(1)通過預先生成的token的頻率對所有候選token進行由高到低排序,可以確保最常見的和最重要的片段被優先考慮,提高粉刺效率。
81、(2)用概率篩選所有token,保留在最優先物算法片計劃中,有效減少了不必要的片段存儲和處理,提升計算效率。
82、(3)將頻率相同的token進行時間分片處理,可以確保在相同條件下,更多樣化的片段組合被用于模型訓練,增加模型的魯棒性和泛化能力。
83、7.本發明采用刪除“低頻片段”的方法優化活性分子片斷庫,即“低頻掩蔽”方法,其優點在于:(1)通過低頻掩蔽的方法減少噪聲和稀疏數據,低頻片段在數據中出現次數較少,可能包含更多的噪聲和隨機性。通過掩蔽低頻片段,可以減少噪聲干擾,提高篩選結果的可靠性和準確性。
84、(2)低頻掩蔽可以減少需要處理的片段數量,使篩選過程更為高效。這有助于縮短計算時間和降低計算資源的消耗,從而加速藥物發現過程。
85、(3)掩蔽低頻片段后,篩選過程可以更多地關注那些高頻且重要的片段。這些片段在數據中出現較多,更有可能具有生物活性或關鍵功能,能夠提高篩選結果的質量。
86、8.本發明采用全局和局部相互作用關系融合的親和力預測方法,其優點在于:
87、(1)通過計算片段間的相互作用,可以更精確地分析蛋白質與分子之間的結合模式。這種精細化的分析能夠捕捉到整體分子模型中可能被忽略的細微相互作用。
88、(2)片段化方法能夠識別和評估單個片段的貢獻,聚合這些局部相互作用來得到整體的結合能量,從而提高了蛋白-分子親和力預測的準確性。
89、(3)通過分析片段與蛋白質特定區域的相互作用,可以更好地理解哪些片段是關鍵的結合元件。這種方法有助于揭示分子設計的基本原理,使得模型具有更好的解釋性。
- 還沒有人留言評論。精彩留言會獲得點贊!