一種蟾毒配基納米晶體及其制備方法與流程
本發明屬于醫藥技術領域,特別是涉及一種蟾毒配基納米晶體及其制備方法。
背景技術:
蟾酥bufonisvenenum源于《本草衍義》,為我國傳統中藥。《中華人民共和國藥典》(2015年版,一部)明確記載了蟾酥是其基源動物蟾蜍科動物中華大蟾蜍bufobufogargarzanscantor或黒眶蟾蜍bufomelanostictusschneider的耳后腺及皮膚腺分泌的白色漿液,經加工干燥而成。蟾酥又名蟾蜍眉酥《日華子本草》、蟾蜍眉脂《藥性論》、癩蛤蟆漿《新疆藥材》、蛤蟆《中藥材手冊》、蛤蟆酥《山東中藥》,其性味辛、溫,有毒;有解毒、止痛、開竅醒神、消腫等功能,主要用于治療癰疽療瘡、咽喉腫痛、中暑神昏、腹痛吐瀉等病癥。
蟾酥含有多種化學成分,其中蟾酥甾類和吲哚生物堿類是其主要活性成分。研究表明,蟾酥抗癌的有效成分主要是蟾酥甾類成分中的蟾毒配基類化合物,其中蟾毒靈、華蟾毒配基和脂蟾毒配基是蟾毒配基中含量較高、活性較強的三個成分,不僅具有抗腫瘤作用,而且還能提高機體免疫力,減輕化療的毒副反應,降低癌痛發生率,增強機體對放療、化療的耐受性,但其水溶性極差,因此需通過相應的制劑工藝提高藥物的溶出速率或表觀溶解度,達到提高其生物利用度的目的。
目前,為了提高蟾酥的生物利用度,研究者采用了諸多方法,如以下文獻所述:
中國發明專利《蟾酥滴丸及其制備方法》(公開號cn1660141a)公開了一種蟾酥滴丸制劑,是利用表面活性劑為基質,與白花蛇舌草的有效成分一起制成固體分散劑,以加快藥物的溶解而提高其生物利用度。但是,該滴丸制劑中所含蟾酥有效成分為蟾酥的乙醇提取物,成分較復雜,并不能很好的保證蟾酥臨床應用的安全性。
中國發明專利《蟾酥提取物在制備治療人腦膠質瘤藥物中的應用》(公開號cn105687251a)公開了蟾毒靈、華蟾酥毒基和脂蟾毒配基的含量大于90%的蟾酥提取物在制備治療人腦膠質瘤藥物中的應用,其中涉及了蟾毒配基脂質納米粒、蟾毒配基納米脂質體、蟾毒配基亞微乳、蟾毒配基納米囊、蟾毒配基納米球、蟾毒配基納米乳或蟾毒配基聚合物膠束,并與蟾毒配基溶液劑進行了比較,結果證實了蟾毒配基納米制劑達到了增效減毒的目的,但這些制劑制備工藝復雜,載藥量較低,大多穩定性較差。
中國發明專利《基于蟾酥提取物的抗腦膠質瘤藥物及其制備方法》(公開號cn105477020a)公開了一種蟾毒配基亞微乳及蟾毒配基納米粒制劑,取得了較理想的抗腦膠質瘤效果,但該蟾毒配基亞微乳和納米粒均存在載藥量低、制備工藝復雜等缺點。
中國發明專利《一種蟾酥靶向脂質體及其制備方法和應用》(公開號cn105561304a)公開了一種蟾酥靶向脂質體,該蟾酥靶向脂質體能夠有針對性的對癌細胞進行抑制同時減小了其毒副作用,由于該脂質體為注射劑,制備工藝復雜且不能消除其在注射時蟾毒配基對血管的刺激性,導致患者順應性差。
綜上所述,上述現有技術方法還沒有達到有效改善蟾毒配基口服生物利用度的目的,特別是實現三種成分同步而快速地溶出,目前仍需要一種優良的方法以進一步改善蟾毒配基的溶出速率而提高其生物利用度。
技術實現要素:
本發明的目的就在于克服上述現有技術的缺陷,提供一種可實現蟾毒配基中蟾毒靈、華蟾毒配基和脂蟾毒配基同步而快速溶出,從而提高蟾毒配基生物利用度,且穩定性好,制備工藝簡單的蟾毒配基納米晶體。
本發明的另一目的是提供上述蟾毒配基納米晶體的制備工藝。
為實現上述發明目的所采取的技術方案為:
一種蟾毒配基納米晶體,其特征在于該蟾毒配基納米晶體是將蟾毒配基、電荷穩定劑、空間穩定劑和純化水混合后經納米化處理后所得,其載藥量為20%~90%,粒徑分布范圍為30~1000nm。
所述蟾毒配基納米晶體中蟾毒配基的總濃度為1mg/ml~500mg/ml;電荷穩定劑的質量體積濃度為0.01%~2.0%;空間立體穩定劑的質量體積濃度為0.01%~5.0%。
所述電荷穩定劑的質量體積濃度為0.10%~0.22%;空間立體穩定劑的質量體積濃度為0.10%~0.30%。
所述蟾毒配基是由蟾毒靈、華蟾毒配基或/和脂蟾毒配基組成的無定形態或微晶態的物質,且其總純度為20~98%,其中由蟾毒靈、華蟾毒配基和脂蟾毒配基三種物質組成時,蟾毒靈占4~15%,華蟾毒配基占6~30%,脂蟾毒配基占10~53%。
所述電荷穩定劑為十二烷基硫酸鈉、泊洛沙姆188、泊洛沙姆407、大豆卵磷脂、海藻糖和殼聚糖中的一種或幾種。
所述空間穩定劑為羥丙基甲基纖維素e5、羧甲基纖維素鈉、交聯羧甲基纖維素鈉、聚維酮k30、羥丙基纖維素和交聯羧甲基淀粉鈉中的一種或幾種。
所述蟾毒配基納米晶體的粒徑范圍為30~200nm,電位值范圍為-58~30mv。
所述蟾毒配基納米晶體經表面修飾或者包衣、干燥處理后,與適宜藥用輔料混制成藥劑學上可接受的各種劑型。
所述表面裝飾是指利用α-生育酚琥珀酸聚乙二醇酯、殼聚糖及其衍生物以物理和/或化學的方式改變蟾毒配基納米晶體的表面結構和狀態。
所述劑型為注射劑、片劑、口服液、微丸或者膠囊劑。
所述納米化處理是指采用濕法研磨法或者高壓均質法。
上述蟾毒配基納米晶體的制備方法,其特征在于其工藝步驟為:
1)將電荷穩定劑、空間穩定劑和純化水充分混合,形成穩定劑溶液a;
2)將蟾毒配基加入到上述穩定劑溶液a中,通過磁力攪拌使藥物混懸于其中,得蟾毒配基粗混懸液b;
3)將上述所得蟾毒配基粗混懸液b轉移至納米研磨機/高壓均質機中進行研磨/均質而得到蟾毒配基納米晶體。
所述納米研磨機為循環式納米研磨機,所用研磨珠的粒度為0.2~0.8mm,材質為玻璃、釔穩定的氧化鋯或聚苯乙烯衍生物的聚合物。
所述研磨過程中,研磨時間控制在15~60min,研磨轉速控制在1000~6000rpm。
所述高壓均質機為超高壓納米均質機,均質壓力為50~200mpa、均質溫度為5~60℃,均質次數為2~20次。
本發明具有以下技術優勢:
1、本發明采用電荷穩定劑、空間穩定劑與蟾毒配基混合制備蟾毒配基納米晶體,充分利用電荷穩定劑所具有的1)提供電荷斥力或與藥物分子以氫鍵等方式結合而減弱藥物粒子的聚集,2)增加藥物的溶解度,3)抑制藥物在分散、破碎過程中產生泡沫,從而增加藥物與藥物、藥物與研磨腔壁、藥物與研磨介質間的碰撞作用等特性;以及空間穩定劑所具有的1)增大空間位阻以防止藥物粒子聚集,2)在一定時間內可維持藥物分子以過飽和狀態存在,3)加快由該納米晶體所制備的其他固體制劑的崩解以利于藥物快速溶出等特性,將由表面活性劑或聚合物構成的電荷穩定劑及空間穩定劑以物理吸附或氫鍵作用等方式包覆于蟾毒配基粒子表面,在阻止藥物聚集的同時避免了藥物分子與外界環境的接觸,最終實現了制備效率和產率的提高,蟾毒配基中三種成分的快速而同步地溶出,蟾毒配基納米晶體物理或/及化學穩定性的提高等等優勢,實驗表明:蟾毒配基納米晶體在人工腸液中三種成分可按最初的質量比同步溶出,且溶出速率比蟾毒配基普通混懸劑提高了約1~10倍;在室溫和低溫條件下儲存兩個月,納米晶體粒徑和藥物含量均無明顯變化。
2、本發明采用納米研磨技術對蟾毒配基進行處理,研磨過程中,藥物粒子之間以及藥物粒子與研磨介質、器壁間在高速剪切力、撞擊力、摩擦力等作用下,將碰撞機械能轉化為混懸液分子勢能,使得藥物粒徑減小,比表面積增大,達到粒度均一的納米晶體;同時通過藥物與穩定劑的共研磨改善了難溶性藥物蟾毒配基的體外溶出特性,并能充分的防止藥物粒子的聚集,保證了混懸液的穩定性。
3、本發明采用高壓均質機對蟾毒配基進行處理,均質過程中,藥物顆粒在空穴、湍流、剪切、撞擊力等共同作用下被粉碎成納米顆粒,穩定劑同時也受此作用形成單分子界面,進而對藥物納米顆粒進行包裹,有效阻止了藥物顆粒的聚集,采用此法制得的納米晶體粒徑均勻,穩定性好。
4、本發明采用的制備工藝簡單,制得的納米晶體載藥量高,穩定性好,可批量化生產。
綜上所述,本發明的蟾毒配基納米晶體同步提高了蟾毒配基中三種成分的溶出速率,載藥量高,穩定性好,為蟾毒配基的其他制劑提供了良好的中間體,且制備工藝簡單、高效、安全,適應工業化生產。
附圖說明
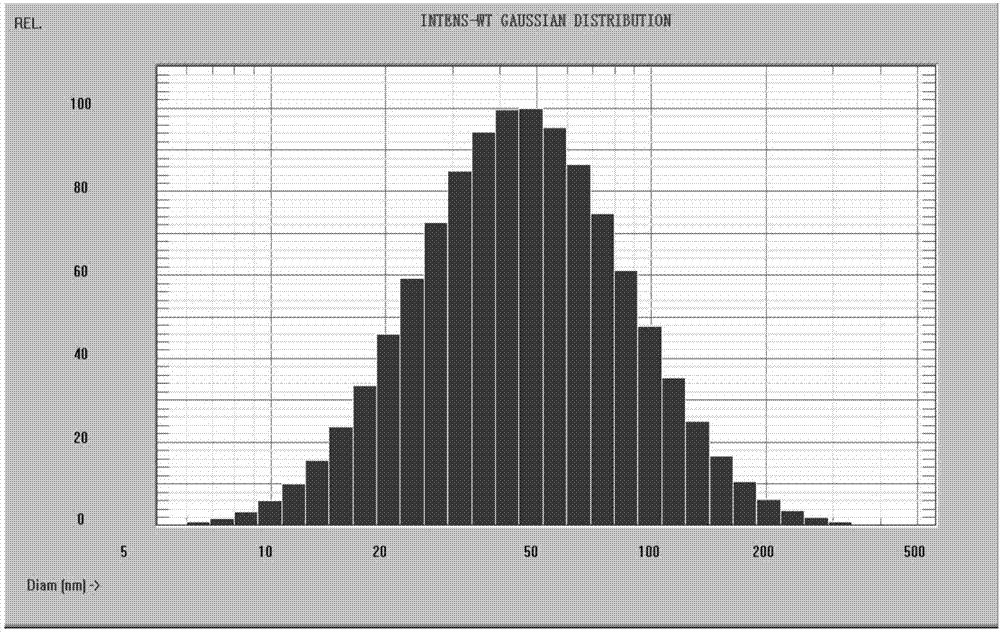
圖1為本發明蟾毒配基納米晶體的粒徑分布圖。
圖2為本發明蟾毒配基納米晶體的透射電鏡圖。
圖3為本發明蟾毒配基納米晶體在人工腸液中(ph=6.8)的溶出曲線圖。
圖4為本發明蟾毒配基納米晶體在純化水中的溶出曲線圖。
圖5為本發明蟾毒配基納米晶體在醋酸緩沖液中(ph=4.5)的溶出曲線圖。
圖6為本發明蟾毒配基納米晶體的紅外光譜圖(a為十二烷基硫酸鈉,b為交聯羧甲基纖維素鈉,c為蟾毒配基原料藥,d為物理混合物,e為納米晶體)。
圖7為本發明蟾毒配基納米晶體的粉末x射線衍射圖(a為十二烷基硫酸鈉,b為交聯羧甲淀粉鈉,c為蟾毒配基原料藥,d為物理混合物,e為納米晶體)。
具體實施方法
下面用實例予以說明本發明,應該理解的是,實例是用于說明本發明而不是對本發明的限制。本發明的范圍與核心內容依據權利要求書加以確定。
下列實施例中,蟾毒配基是由蟾毒靈、華蟾毒配基和脂蟾毒配基三種成分組成的無定形態或微晶態的混合物,總純度為20~98%,其中蟾毒靈約占4~15%,華蟾毒配基約占6~30%,脂蟾毒配基約占10~53%。
實施例1:一種蟾毒配基納米晶體的制備
稱取十二烷基硫酸鈉0.08g,交聯羧甲基纖維素鈉0.05g,加入50ml蒸餾水中,使其充分溶解溶脹,在磁力攪拌下加入0.5g蟾毒配基原料藥,磁力攪拌30分鐘,得粗混懸液。然后將此粗混懸液轉移至納米研磨機中進行研磨。研磨條件為:釔穩定的氧化鋯研磨珠粒徑為0.6-0.8mm,攪拌軸轉速為2000rpm。30分鐘后取樣,測得粒徑為30.2nm,電位為-47.9mv。
實施例2:一種蟾毒配基納米晶體的制備
稱取泊洛沙姆1880.05g,羥丙甲纖維素0.5g,加入50ml蒸餾水中,使其充分溶解溶脹,在磁力攪拌下加入0.8g蟾毒配基原料藥,攪拌30分鐘,得粗混懸液。然后將此粗混懸液轉移至納米研磨機中進行研磨。研磨條件為:聚苯乙烯聚合物研磨珠粒徑為0.2-0.4mm,攪拌軸轉速為1000rpm。60分鐘后取樣,測粒徑為238.5nm,電位為-5.9mv。
實施例3:一種蟾毒配基納米晶體的制備
稱取大豆卵磷脂0.1g,聚維酮k300.05g,海藻糖0.02g,加入50ml蒸餾水中,使其充分溶解溶脹,在磁力攪拌下加入0.05g蟾毒配基原料藥,超聲30分鐘,得粗混懸液。然后將此粗混懸液轉移至納米研磨機中進行研磨。研磨條件為:玻璃研磨珠粒徑為0.3-0.5mm,攪拌軸轉速為6000rpm。15分鐘后取樣,測粒徑為973.8nm,電位為-20.9mv。
實施例4:一種蟾毒配基納米晶體的制備
稱取殼聚糖0.08g,交聯羧甲基纖維素鈉0.15g,加入50ml蒸餾水中,使其充分溶解溶脹,在磁力攪拌下加入0.5g蟾毒配基原料藥,超聲混懸20分鐘,得粗混懸液。然后將此粗混懸液轉移至高壓均質機中進行均質。均質條件為:壓力為200mpa,溫度為60℃,循環2次,測粒徑為720.8nm,電位為-20.9mv。
實施例5:一種蟾毒配基納米晶體的制備
稱取泊洛沙姆4070.1g,羥丙基纖維素0.05g,交聯羧甲淀粉鈉0.1g,加入50ml蒸餾水中,使其充分溶解溶脹,在磁力攪拌下加入0.6g蟾毒配基原料藥,攪拌20分鐘,得粗混懸液。然后將此粗混懸液轉移至高壓均質機中進行均質。均質條件為:壓力位50mpa,溫度為5℃,循環20次,測粒徑為608.8nm,電位為-2.9mv。
實施例6:蟾毒配基納米晶體的表面修飾:
稱取0.1g的羥丙基三甲基氯化銨殼聚糖(hacc),將其溶解于100ml的蒸餾水中得0.1%的hacc溶液。將此hacc溶液于探頭超聲作用下緩慢加入到實施例1所制得的蟾毒配基納米晶體中(納米晶體與修飾液的體積比為1:4),然后磁力攪拌3小時,測定經修飾的納米晶體的粒徑及電位。
實施例7:蟾毒配基納米晶體冷凍干燥粉末的制備:
取制備的蟾毒配基納米晶體新鮮樣品,加入蔗糖作為凍干保護劑,其濃度為1%(m/v),然后在-70℃預凍12小時,然后轉移至冷凍干燥機中進行凍干,冷阱溫度為-60℃,冷凍干燥時間為24小時,得其凍干粉。
實施例8:溶出度測定:
對蟾毒配基原料藥和實施例2制得的蟾毒配基納米晶體冷凍干燥粉末進行溶出度測定,方法如下:
采用《中國藥典》2015版四部溶出度與釋放度測定法(第二法),分別以900ml的純化水、ph6.8磷酸鹽緩沖液、ph4.5醋酸緩沖液作為漏槽條件下的溶出介質。溶出條件和實驗操作如下:量取經過脫氣處理的溶出介質900ml,注入1000ml溶出杯中,槳法,轉速為50rpm,溫度為(37±0.5)℃。待溫度穩定后,分別向各溶出杯中投入樣品,分別于0,5,10,15,30,45,60,90,120min取樣5ml,用0.22μm的濾膜過濾,同時補加5ml新介質,取續濾液作為供試品溶液。另取蟾毒靈、華蟾毒配基對照品各3mg,脂蟾毒配基對照品約5mg,精密稱定,溶于10ml甲醇后,取其中1ml以溶出介質定量稀釋至10ml,作為對照品溶液。采用高效液相色譜法進行含量測定,計算蟾毒配基原料藥和蟾毒配基納米晶體凍干粉末中蟾毒靈、華蟾毒配基和脂蟾毒配基的溶出度,結果見圖3。
- 還沒有人留言評論。精彩留言會獲得點贊!