控釋氫可酮制劑的制作方法
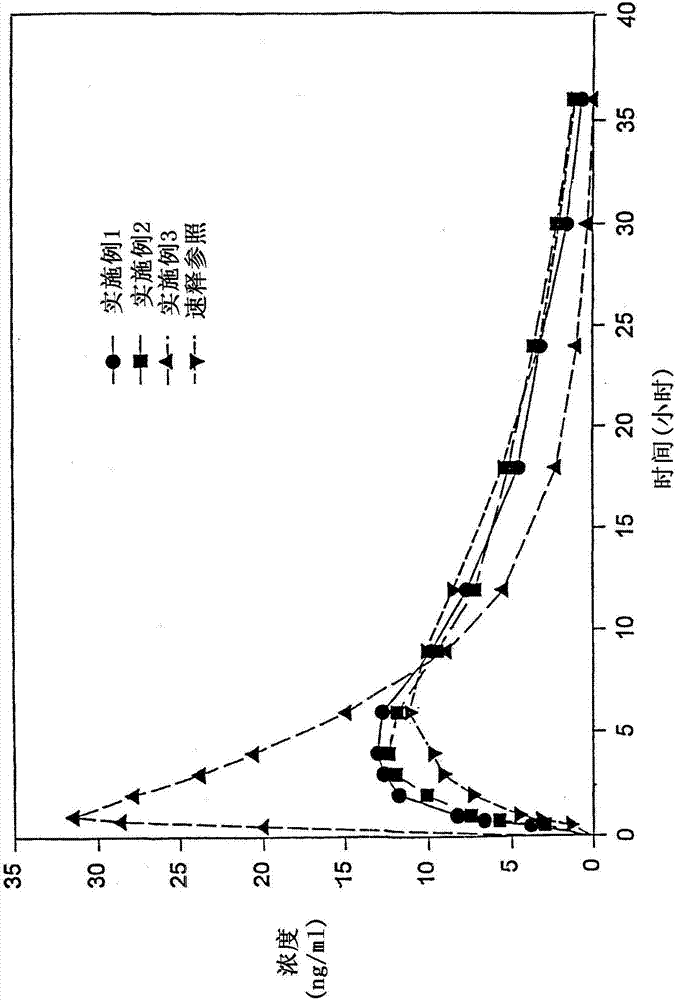
發明背景由于藥物治療難以治愈疼痛,尤其是慢性疼痛,所以,阿片類鎮痛劑是理想的控釋給藥制藥物。本發明是關于一種用于治療疼痛的固體口服控釋劑型。所有控釋(緩釋)制劑的目的都是在給藥后能比速釋劑型更持久地提供藥粒作用。這樣的長效與速釋制劑的短效相比具有許多治療上的好處。這樣可以在不打擾患者睡眠的前提下繼續治療,例如在治療中等至劇烈疼痛患者(例如術后患者,癌癥患者等)時,或對于醒時會發生偏頭痛的患者來說,或對于亟需睡夢的衰弱患者來說,這非常重要。速效藥物治療除非精心定時給藥以維持藥物穩定的有效血漿濃度,由于對化合物的迅速吸收、全身性排泄和代謝滅活,活性藥物的血漿水平會出現峰谷,因此在對患者療效的維持上造成問題。長效藥物的另一優點是提高了患者的順應性,因為它避免了患者因遺忘而遺漏給藥。制備可在人和動物口服后緩慢釋放所含藥學活性物質的組合物在制藥業中是已知技術。此類緩釋組合物被用來使得藥物吸收延遲至到達消化道特定部位時才發生。與傳統速釋劑型給藥相比,這種藥物在消化道內的控釋可在更長時間內將藥物的血液濃度維持在要求的水平。有關制備和使用從載體中緩慢釋放活性化合物的組合物的現有技術都涉及活性物質向消化道生理液中的釋放。然而,都知道,單獨存在于胃腸液內的活性物質本身不能確保其生物利用度。為了被吸收,活性藥物必需呈溶液狀態。單位劑型釋出一定量活性物質所需的時間可在標準條件下測定,表示為特定時間內由該單位劑型釋放出的活性藥物量比例。生理腸胃液是測定溶出時間的介質。現有技術認可許多試驗可測定藥物組合物的溶出時間,這些試驗在世界各地的各類正式藥典中都有所論述。雖然有許多因素影響著藥物從其載體中的溶出,但試驗測得活性藥物從具體組合物中溶出的時間相對穩定并具有重復性。影響溶出時間的因素包括藥物接觸溶劑介質的表面積,溶液的ph,藥物在具體溶劑中的溶解度,和溶劑中物質的飽和濃度的驅動力。因此,當活性藥物在組織部位被吸收而從溶劑中消除時,其溶出濃度動態地保持著穩態。在生理條件下,溶質的飽和水平得到劑型中儲量的補充,因而在溶劑中維持著相對均一和恒定的溶出濃度,以實現穩定吸收。胃腸道組織吸收部位的跨組織運輸受到膜兩側donnan滲透平衡力的影響,因為,驅動力的方向就是膜兩側活性物質的濃度差,即,溶于胃腸液內和血液內活性物質量的差異。由于血液濃度因稀釋、循環改變、組織儲存、代謝轉化和全身性排泄而不斷改變,所以,活性物質的流向是從胃腸道進入血液。已有多種技術被用于制備控釋劑型。具體地說,包衣丸粒、片劑和膠囊通過其包衣的選擇性崩解或通過與特定基質混合以影響藥物釋放來緩慢釋放活性藥物,這是本領域的已知技術。某些控釋制劑可實現單劑活性化合物給藥后在預定時段順次釋放。專利文獻中記載的控釋阿片類制劑例如:美國專利4,990,341和4,844,909(goldie等),此兩專利均已轉讓給了本發明的受讓人,其中,經37℃,900ml緩沖液(ph1.6-7.2),100rpm的usp槳法或轉籃法測定,所述氫嗎啡酮組合物劑型的體外溶出率為:1小時后為12.5-42.5wt%氫嗎啡酮,2小時后為25-55wt%,4小時后為45-75wt%,6小時后為55-85wt%,體外釋放率在ph1.6-7.2區間內與ph無關,而且,體內氫嗎啡酮血漿濃度峰值出現在給藥后2-4小時之間。上述氫嗎啡酮制劑緩解疼痛至少可達12小時。目前,非常需要可用于治療中度疼痛的其他阿片類鎮痛劑的緩釋劑型。而且,非常需要其藥物動力學特性可提供最佳鎮痛療效的控釋劑型。發明概述本發明目的之一是顯著提高對中度疼痛患者鎮痛療效。本發明目的還在于提供可顯著改善鎮痛效率和效果的可生物利用的氫可酮制劑。本發明目的還在于提供可生物利用的氫可酮控釋制劑,其療鎮痛效果效持續時間顯著長于速釋氫可酮制劑,但起效迅速。本發明的目的還在于提供適合每日兩次給藥的口服控釋阿片類制劑,它起效早,在給藥間期內達到最高濃度后具有相對平緩的血漿曲線,阿片樣物質血漿水平的c12/cmax之比為0.55-0.85,并可有效地緩解患者的疼痛。在另一些實施方式中,所述劑型的c12/cmax之比為0.65-0.75。上述及其他目的可藉本發明的特征得以實現,在某些實施方式中,提供了一種固體口服控釋劑型,其中包含鎮痛有效量的氫可酮或其藥學上認可的鹽和足量的控釋材料,以使該劑型適合每日兩次給藥,該劑型在一次給予人患者后,體內氫可酮血漿濃度峰值出現時間約為2-8小時后(tmax),達到最高濃度后的c12/cmax之比為0.55-0.85。在部分優選實施方式中,通過usp轉籃法測定:37℃,先在700ml模擬胃液(sgf)中,100rpm,55分鐘,然后轉入900ml模擬腸液(sif)中,37℃,測得所述控釋劑型1小時后的氫可酮或其鹽體外釋放率為18-42.5wt%。在部分優選實施方式中,通過usp轉籃法測定:37℃,900mlph1.2-7.5緩沖液中,100rpm,所述氫可酮劑型的氫可酮或其藥學上認可的鹽的體外溶出率為:2小時后約為25-65wt%,4小時后為45-85wt%,8小時后超過60wt%。雖然,根據要求,體外釋放率可以是非ph依賴性的,也可以是ph依賴性的,在本發明優選實施方式中,氫可酮的釋放是非ph依賴性的。在部分優選實施方式中,提供了一種包含治療有效量的氫可酮的控釋劑型,該劑型在給藥后12小時的氫可酮血漿濃度至少為5-6ng/ml,給藥后2-8小時,至少為8ng/ml。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,該劑型的氫可酮cmax低于等劑量速釋氫可酮參比制劑(例如)cmax的50%,并可在12小時的給藥間期內有效鎮痛。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,該劑型達到80%cmax所需的時間約為等劑量速釋氫可酮參比制劑(例如)的90-150%,以約90-110%為佳。較好的是,控釋劑型達到80%氫可酮cmax所需的時間為0.5-1.5小時左右,最好是0.8-1.2小時。另一些實施方式中,控釋劑型達到80%氫可酮cmax所需的時間為0.75-2.0小時左右,最好是0.9-1.5小時。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,該劑型達到90%cmax所需的時間約為等劑量速釋氫可酮參比制劑(例如)的150-400%,以約150-250%為佳。較好的是,控釋劑型達到90%氫可酮cmax所需的時間為1.5-2.5小時左右,最好是1.8-2.2小時。另一些實施方式中,控釋劑型達到90%氫可酮cmax所需的時間為1.5-4.0小時左右,最好是1.8-2.5小時。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,該劑型將血漿濃度維持在80%cmax的時間可達約0.5-10小時,以1-9小時或4-8小時為佳。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,該劑型將血漿濃度維持在90%cmax的時間可達約1-6.5小時,以2-5小時或2-6.5小時為佳。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,以含15mg氫可酮二酒石酸鹽的口服劑型為基準,該劑型從給藥到tmax期間的平均體內吸收速度為1.5-5mg/h,從tmax到給藥間期末的平均吸收速度低于0.5mg/h。較好的是,以含15mg氫可酮二酒石酸鹽的口服劑型為基準,該劑型從給藥到tmax期間的平均體內吸收速度約為2-4mg/h,從tmax到12小時給藥間期末的平均吸收速度約為0.08-0.4mg/h。在本發明另一些優選實施方式中,提供了一種日用兩次的氫可酮口服控釋劑型,從tmax到給藥后12小時期間內,該劑型的吸收速度是消除速度的約55-85%。較好的是,本發明上述及其他實施方式的tmax時刻比等劑量速釋氫可酮參比制劑遲3-4倍。較好的是,所述緩釋制劑的tmax出現在口服給藥后約2-8小時,約3-7小時,或4-6小時。本發明還涉及如下氫可酮制劑:其氫可酮cmax低于等劑量速釋參比產品的約50%,較好的是低于約40%。例如,本發明出人意料的發現,當氫可酮配在美國專利4,861,598和4,970,075所述傳遞系統中時,所述系統的氫可酮cmax相對于速釋參比產品的百分比低于配制于相同傳遞系統內的羥考酮。雖然羥考酮和氫可酮控釋制劑的體外溶出情況相似,但上述現象卻十分明顯。用美國專利4,861,598和4,970,075所述傳遞系統配制本發明時,所述系統cmax相對于速釋參比產品的百分比低于50%,優選實施方式低于40%,羥考酮則高于50%。就本發明目的而言,“氫可酮”包括氫可酮游離堿,及其藥學上認可的鹽和復合物。“usp槳法或轉籃法”即us藥典xxii(1990)中記載的方法。就本發明目的而言,“ph依賴性”表示隨周圍ph而改變的特性(例如溶出)。就本發明目的而言,“非ph依賴性”表示不受周圍ph影響的特性(例如溶出)。就本發明目的而言,“生物利用度”表示單位劑型中藥物(例如氫可酮)被吸收的程度。就本發明目的而言,“控釋”表示藥物(例如氫可酮)的釋放速度令血液(如血漿)濃度在至少約12小時內維持在療效范圍內,但低于毒性濃度。“cmax”表示給藥間期內達到的最高血漿濃度。“tmax”表示達到最高血漿濃度(cmax)所需的時間。“t1/2(吸收)”表示半數可吸收劑量阿片樣物質轉運進入血漿所需的時間。“穩態”表示給定藥物達到的血漿濃度通過連續給藥而維持在該藥物最低療效濃度以上、最低毒性血漿濃度以下。就阿片類鎮痛劑而言,最低療效濃度在一定程度上可根據特定患者所獲得的疼痛緩解程度來確定。醫藥領域技術人員都知道,疼痛程度具有很強的主觀性,而且患者間個體差異較大。就本發明目的而言,“維持治療”和“長期治療”指在患者經阿片類鎮痛劑給藥達到上述穩態后給予患者的藥物治療。阿片類例如氫可酮的“最低鎮痛有效濃度”或“meac”非常難以確定。然而,通常有一個氫可酮最低鎮痛有效血漿濃度,該濃度以下沒有鎮痛效果。雖然在例如氫可酮血漿水平與鎮痛效果之間存在著間接關聯,但較高較持久的血漿水平通常與較佳的鎮痛效果相關。氫可酮血漿水平達到最高的時刻與藥物效果達到最大的時刻之間有一段滯后期。所有阿片類鎮痛劑治療疼痛都存在這一現象。“平均共振時間(meanreasonancetime”(mrt)定義為藥物分子在體內的平均停留時間。該算得值與吸收、分布和消除有關,一定程度上取決于含活性成分的劑型。本發明中,除非另外說明,“一名患者”表示所作的論述(或權利要求)是單獨一名患者的藥物動力學情況。“患者群”表示表示所作的論述(或權利要求)是至少兩名患者的藥物動力學情況。“突破性疼痛(breakthroughpain)”指盡管該患者正在接受一般有效量的含氫嗎啡酮的本發明緩釋固體口服劑型給藥而仍然經受的疼痛,。“援救”指給予正在經受突破性疼痛的患者的鎮痛劑劑量。“有效鎮痛”指醫生對患者接受鎮痛治療的反應的客觀評價和接受該治療的患者對治療的主觀評價。本領域熟練技術人員可以看出,有效鎮痛取決于許多因素,其中包括患者的個體差異。就本發明目的而言,“氫可酮速釋參比制劑”是等劑量的(購自ucbpharma,inc)中的氫可酮,或其他氫可酮或其藥學上認可的鹽的速釋藥物產品中的氫可酮。本發明中,所述的控釋制劑和速釋制劑都是劑量比例性的。此類制劑中,藥物動力學參數(例如auc和cmax)從一個劑量強度向另一劑量強度的提高呈線性。所以,某一特定劑量的藥物動力學參數可由該制劑其他劑量的參數推導得出。本發明中,除非另作說明,所述的藥物動力學參數都以一名患者單獨一次給予氫可酮制劑為基礎。基于患者群的藥物動力學參數將以“平均”數表示。“第一次給藥”指在治療開始時向一名患者或患者群的一次給藥。出人意料的是,本發明的控釋口服固體劑型可節省阿片樣物質的量。本發明控釋口服固體劑型的日給藥量可大大低于常規速釋產品,但在鎮痛效果上卻沒有差異。與常規速釋產品相比,在相當的日劑量上,采用本發明的控釋口服固體劑型可獲得更好的效果。附圖簡述附圖用于說明本發明的實施方式而不是象權利要求那樣限定本發明的范圍。圖1顯示實施例1,2,3和一種等劑量速釋氫可酮的平均氫可酮血漿濃度。圖2顯示實施例1,2和3的平均氫可酮血漿濃度,與之相比的是按照實施例4方法制成的控釋羥考酮樣品,按照實施例5方法制成的控釋嗎啡樣品。圖3顯示實施例1,2,3和一種等劑量速釋氫可酮在不同時間的氫可酮吸收百分比。詳細描述本發明的上述實施方式可用多種本領域已知的控釋制劑來實施。例如美國專利4,861,598和4,970,075所述的控釋劑型。在本發明部分實施方式中,制劑中包含速釋劑型的有效量的阿片樣物質。所含速釋劑型阿片樣物質的量可有效縮短達到阿片樣物質在血液(例如血漿)中最高濃度所需的時間,使得tmax縮短至例如2-5小時或2-4小時。已發現,在單位劑型中包含上述有效量的速釋阿片樣物質可明顯緩解患者較強烈的疼痛。在此類實施方式中,可將所述有效量的速釋阿片樣物質包涂在本發明基質之上。例如,在由控釋包衣實現阿片樣物質從制劑中延時釋放時,可將速釋層包在控釋包衣之外。另一方面,當阿片樣物質摻和在控釋基質中時,可將速釋層包在基質表面上。如果是將含有效單位劑量阿片樣物質的多份緩釋基質(例如丸粒、球粒、小珠等的多顆粒系統)包含在一個硬明膠膠囊內,可將阿片劑量的速釋部分以粉末或顆粒形式摻入明膠膠囊。或者,明膠膠囊本身可包以阿片樣物質速釋層包衣。本領域技術人員知道,還有其他方式可將阿片樣物質的速釋部分加入單位劑量。這些方式應視為包含在后文權利要求中。本發明阿片樣物質劑型的優點之一是,在基本達到治療濃度的同時,伴生副作用的強度和/或程度沒有明顯增強,所述副作用例如常伴隨阿片樣物質高血液濃度發生的惡心、嘔吐或嗜睡。還有證據表明,采用本發明劑型可降低藥物成癮的危險性。活性物質本發明控釋口服劑型宜包含約0.5-1250mg氫可酮或等量的其藥學上認可的鹽,在優選實施方式中,可以是約5-60mg,例如15mg。氫可酮合適的藥學上認可的鹽包括:二酒石酸氫可酮,二酒石酸氫可酮水合物,鹽酸氫可酮,對甲苯磺酸氫可酮,磷酸氫可酮,氫可酮縮氨基硫脲,硫酸氫可酮,三氟乙酸氫可酮,氫可酮二倍半水合物,五氟丙酸氫可酮,氫可酮對硝基苯腙,氫可酮o-甲基肟,氫可酮縮氨基脲,氫溴酸氫可酮,粘酸氫可酮,油酸氫可酮,磷酸二氫氫可酮,磷酸氫氫可酮,氫可酮無機酸鹽,氫可酮有機酸鹽,乙酸氫可酮三水合物,二(七氟丁酸)氫可酮,二(甲基氨基甲酸)氫可酮,二(五氟丙酸)氫可酮,二(吡啶羧酸)氫可酮,二(三氟乙酸)氫可酮,鹽酸氫可酮,和硫酸氫可酮五水合物。本發明優選二酒石酸氫可酮。本發明劑型還可以包含一種或多種其他藥物,它們與本發明的氫可酮鎮痛劑或有或沒有協同作用。此類藥物的例子包括:非甾體類消炎藥,如布洛芬,雙氯芬酸,萘普生,苯洛芬,氟比洛芬,非諾洛芬,flubfen,酮洛芬,吲哚洛芬,吡洛芬,卡洛芬,丙嗪,普拉洛芬,莫拉洛芬,三氧洛芬,舒洛芬,氨基洛芬,噻洛芬酸,氟洛芬,布氯酸,吲哚美辛,舒林酸,托美丁,佐美酸,硫平酸,齊多美辛,阿西美辛,芬替酸,環氯茚酸,奧西平酸,甲芬那酸,甲氯芬那酸,氟芬那酸,尼氟滅酸,托芬那酸,二氟尼柳,氟苯柳,吡羅昔康,舒多昔康或依索昔康。此類非甾體消炎藥還包括環加氧酶抑制劑,如celecoxib(sc-58635),dup-697,flosulide(cgp-28238),美羅昔康,6-甲氧基-2-萘基乙酸(6-mna),vioxx(mk-966),奈丁美酮(6-mna的前藥),尼美舒利,ns-398,sc-5766,sc-58215和t-614,金剛烷胺(1-氨基金剛烷)和美金剛(3,5-二甲基氨基金剛烷酮),它們的混合物,以及它們藥學上認可的鹽。其他附加藥物包括無毒nmda受體拮抗劑,例如右啡烷,右美沙芬,3-(1-萘基)-5-(膦酰基甲基)-l-苯丙氨酸,3-(1-萘基)-5-(膦酰基甲基)-dl-苯丙氨酸,1-(3,5-二甲基苯基)萘和2-(3,5-二甲基苯基)萘,2sr,4rs-4-(((1h-四唑-5-基)甲基)氧)哌啶-2-羧酸;2sr,4rs-4-((((1h-四唑-5-基)甲基)氧)甲基)哌啶-2-羧酸;e和z2sr-4-(o-(1h-四唑-5-基)甲基)甲酮肟基)哌啶-2-羧酸;2sr,4rs-4-((1h-四唑-5-基)硫)哌啶-2-羧酸;2sr,4rs-4-(5-巰基-1h-四唑-1-基)哌啶-2-羧酸;2sr,4rs-4-(5-巰基-2h-四唑-2-基)哌啶-2-羧酸;2sr,4rs-4-(5-巰基-1h-四唑-1-基)哌啶-2-羧酸;2sr,4rs-4-(5-巰基-2h-四唑-2-基)哌啶-2-羧酸;,2sr,4rs-4-(((1h-四唑-5-基)硫)甲基)哌啶-2-羧酸;2sr,4rs-4-((5-巰基-1h-四唑-1-基)甲基)哌啶-2-羧酸;或2sr,4rs-4-((5-巰基-2h-四唑-2-基)甲基)哌啶-2-羧酸;它們的混合物,以及它們藥學上認可的鹽。可包含在本發明制劑中的其他合適的附加藥物包括:對乙酰氨基酚,阿司匹林,神經活性甾體(例如美國專利申請09/026,520(1998年2月20日申請)中所述)以及其他非阿片類鎮痛劑。例如,如果制劑中包含第二藥物(非阿片類),該藥物在其中的形式可以是控釋劑型也可以是速釋劑型。該附加藥物可與阿片樣物質一同包含在控釋基質中;包含在控釋包衣中;作為另一控釋層或速釋層包括在制劑中;或以粉末、顆粒等形式與本發明基質一同摻和在明膠膠囊中。在本發明部分實施方式中,給藥用的控釋單位劑型氫可酮制劑中含有有效量的速釋劑型氫可酮。該速釋氫可酮的含量可有效縮短達到血液(例如血漿)內氫可酮cmax所需的時間。此類實施方式中,有效量的速釋氫可酮可包在本發明基質之外。例如,如果由一層控釋包衣來實現氫可酮的延遲釋放,則可將速釋層包在控釋包衣之外。另一方面,如果氫可酮包含在控釋基質中,則可將速釋層包在基質之外。如果是將含有效單位劑量阿片樣物質的多份緩釋基質(例如丸粒、球粒、小珠等的多顆粒系統)包含在一個硬明膠膠囊內,可將阿片劑量的速釋部分以粉末或顆粒形式摻入明膠膠囊。或者,明膠膠囊本身可包以阿片樣物質速釋層包衣。本領域技術人員知道,還有其他方式可將速釋氫可酮部分加入單位劑量。這些方式應視為包含在后文權利要求中。已發現,通過在單位劑型中加入有效量的速釋氫可酮,可顯著緩解患者較為嚴重的疼痛。劑型所述控釋劑型可以任選性地包含一種控釋材料,將其與氫可酮一起摻入基質,或將其作為緩釋包衣包在含藥物的基質(所謂“基質包括丸粒、顆粒、球體、片劑和片芯等)之外。根據需要,所述控釋材料可以是疏水性的也可以是親水性的。本發明口服劑型可以是例如顆粒,球粒,丸粒粒(后文統稱“多顆粒”)。可將含量足以在一段時間內有效提供所需劑量阿片樣物質的多顆粒裝入膠囊,或加入其他各種合適的口服固體劑型,例如壓成片劑。另一方面,本發明所述口服劑型可以制成片芯,然后包以控釋包衣,或制成基質中包含藥物、控釋材料和可選性其他成分(例如稀釋劑,粘合劑,色素,潤滑劑等)的片劑。控釋基質制劑在本發明部分實施方式中,控釋制劑是由一種基質(例如基質片劑)制得的,該基質中包含有前述控釋材料。含控釋基質的劑型具有優選范圍內的阿片樣物質體外溶出速度,并具有ph依賴性或非ph依賴性的溶出方式。適合包含在控釋基質內的材料取決于形成基質的方法。口服劑型可包含1-80wt%至少一種親水性或疏水性控釋材料。適合包含在本發明控釋基質中的控釋材料包括但不限于以下親水性和/或疏水性材料,例如樹膠,纖維素醚,丙烯酸樹脂,蛋白質衍生材料,蠟,蟲漆和氫化蓖麻油、氫化植物油等油脂。然而,任何藥學上認可的疏水性或親水性控釋材料只要能實現阿片樣物質的控釋都可用于本發明。優選的控釋聚合物包括烷基纖維素,例如乙基纖維素,丙烯酸和甲基丙烯酸聚合物及共聚物,纖維素醚,尤其是羥烷基纖維素(尤其是羥丙基甲基纖維素)和羧基烷基纖維素。優選的丙烯酸和甲基丙烯酸聚合物及共聚物包括甲基丙烯酸甲酯,甲基丙烯酸甲酯共聚物,甲基丙烯酸乙氧基乙酯,甲基丙烯酸氰基乙酯,甲基丙烯酸氨基烷酯共聚物,聚(丙烯酸),聚(甲基丙烯酸),甲基丙烯酸-烷基胺共聚物,聚(甲基丙烯酸甲酯),聚(甲基丙烯酸)(酐),聚甲基丙烯酸酯,聚丙烯酰胺,聚(甲基丙烯酸酐)和甲基丙烯酸縮水甘油酯共聚物。某些優選實施例將上述控釋材料混合物用于本發明基質中。所述基質還可包含粘合劑。在此類實施方式中,所述粘合劑最好對于氫可酮從控釋基質中的有控釋放具有一定貢獻。優選的疏水性粘合劑是或多或少具有明顯親水和/或疏水傾向的水不溶性物質。較好的是,可用于本發明的疏水性粘合劑其熔點約為30-200℃,以45-90℃為佳。當所述疏水性物質是烴時,其熔點最好在25-90℃之間。在長鏈烴(c8-50)中,優選脂肪醇。本發明口服劑型可含至多80wt%的至少一種可消化長鏈烴。較好的是,所述口服劑型含至多80wt%至少一種聚烷二醇。具體地說,疏水性粘合劑可包含天然或合成蠟,脂肪醇(例如月桂醇,肉豆蔻醇,硬脂醇,鯨蠟醇,或優選的鯨蠟硬脂醇),脂肪酸(包括但不限于脂肪酸酯,脂肪酸甘油酯(單酯,二酯或三酯),氫化脂肪,烴,普通蠟,硬脂酸,硬脂醇和具有烴骨架的疏水性和親水性材料。合適的蠟包括例如峰蠟,糖蠟(glycowax),蓖麻蠟和巴西棕櫚蠟。就本發明目的而言,蠟樣物質指室溫下一般為固體,熔點約為30-100℃的物質。可用于本發明的優選疏水性粘合劑包括可消化的長鏈(c8-50,尤其是c12-40)飽和或不飽和烴,例如脂肪酸,脂肪醇,甘油脂肪酸酯,礦物油和植物油,天然及合成蠟和聚烷二醇。優選熔點約為25-90℃的烴。在長鏈烴粘合劑中,部分實施方式優選脂肪醇。本發明口服劑型可含有至多80wt%至少一種可消化長鏈烴。在部分優選實施方式中,基質制劑中包含兩種或兩種以上疏水性粘合劑的混合物。如果含有另一種疏水性粘合劑,它最好選自天然及合成蠟、脂肪酸、脂肪醇及它們的混合物。其實例包括峰蠟、巴西棕櫚蠟,硬脂酸和硬脂醇。以上并非窮舉。一種具體的優選控釋基質包含至少一種水溶性羥烷基纖維素,至少一種c12-36(c14-22更好)脂肪醇,以及可選的至少一種聚烷二醇。所述羥烷基纖維素優選c1-6羥烷基纖維素,例如羥丙基纖維素,羥丙基甲基纖維素,尤其是羥乙基纖維素。這至少一種羥烷基纖維素在本發明口服劑型中的含量取決于所要求的阿片樣物質準確釋放速度等因素。脂肪醇可以是例如月桂醇、肉豆蔻醇或硬脂醇。然而,在一特別優選的本發明口服劑型實施方式中,所述至少一種脂肪醇是鯨蠟醇或鯨蠟硬脂醇。本發明口服劑型中脂肪醇的含量,如前所述,取決于所要求的阿片樣物質準確釋放速度,還取決于口服劑型中是否含有至少一種聚烷二醇。如果沒有聚烷二醇,口服劑型宜包含20-50wt%脂肪醇;如果含有聚烷二醇,則脂肪醇與聚烷二醇含量之和宜為劑型總量的20-50wt%。在一優選實施方式中,例如至少一種羥烷基纖維素或丙烯酸樹脂與至少一種脂肪醇/聚烷二醇之比在相當程度上決定著阿片樣物質從制劑中釋放的速度。羥烷基纖維素與脂肪醇/聚烷二醇之比為1:2至1:4,優選1:3至1:4。所述聚烷二醇可以是例如聚丙二醇或優選的聚乙二醇。所述至少一種聚烷二醇的數均分子量以1,000-15,000為宜,以1,500-12,000為佳。另一種合適的控釋基質含有烷基纖維素(尤其是乙基纖維素),c12-36脂肪醇和可選的聚烷二醇。除上述組分之外,控釋基質還可包含適量的其他材料,例如制藥業常用的稀釋劑,潤滑劑,粘合劑,造粒助劑,色素,香精和助流劑。為了方便制備本發明固體控釋口服劑型,本發明的另一方面內容是其制備方法,包括將阿片樣物質或其鹽混合到控釋基質中,可如下進行:(a)制作包含至少一種上述疏水性和/或親水性物質(例如水溶性羥烷基纖維素)和氫可酮的顆粒;(b)將至少含一種疏水性和/或親水性物質的顆粒與至少一種c12-36脂肪醇混合;(c)可選的是,對顆粒進行壓制和成形。可用藥物制劑業熟知的各種方法制備上述顆粒。例如,在一優選方法中,通過用水進行羥烷基纖維素/阿片樣物質濕法造粒來形成所述顆粒。在該方法的一種特別優選實施方式中,濕法造粒過程中水的加量是阿片樣物質干重的1.5-5倍,1.75-3倍更好。本發明的基質還可以通過熔體制丸(pellitization)技術來制備。此時,精細粉碎的阿片樣物質與粘合劑(同為顆粒形式)以及可選的其他惰性成分混合,然后通過例如高剪切混合機中的機械作用將混合物制成顆粒(顆粒,球粒)。然后,篩選具有規定大小的丸粒(顆粒,球粒)。粘合劑最好呈顆粒形式,且熔點高于40℃。合適的粘合劑包括例如氫化蓖麻油,氫化植物油,其他氫化脂肪,脂肪醇,脂肪酸酯,脂肪酸甘油酯等。也可通過例如熔體造粒(granulation)或熔體擠塑技術來制備控釋基質。熔體造粒技術一般包括將通常呈固態的疏水性粘合劑(例如蠟)熔化,摻入藥物粉末。為了獲得控釋劑型,可能需要將疏水性控釋材料(例如乙基纖維素或水溶性丙烯酸聚合物)摻入熔融的蠟類疏水性粘合劑中。有關熔體造粒技術制備的控釋制劑可參見美國專利4,861,598,該專利已轉讓給本發明的受讓人。附加的疏水性粘合劑可包含一種或多種水不溶性蠟樣熱塑性物質,這些物質可能混合著另外的疏水性略低的蠟樣熱塑性物質。為了實現控釋,該制劑中的各種蠟樣物質在釋放初期都必需基本上不在胃腸液中降解或溶解。有用的水不溶性蠟樣粘合劑是水中溶解度低于1:5,000(w/w)的那些。除以上組分之外,控釋基質還可根據需要包含適量其他物質,例如制藥業常用的稀釋劑,潤滑劑,粘合劑,造粒助劑,色素,香精和助流劑(glidant),它們的總量約為顆粒總量的50wt%。這些附加材料的量應足以對所需制劑產生所需效果。用來配制口服劑型的藥學上認可的載體和賦形劑的具體例子可參見藥用輔料手冊,americanpharmaceuticalassociation(1986)。制備合適的本發明熔體擠塑型基質包括:將阿片類鎮痛劑與控釋材料,最好還有粘合劑,混合成均勻的混合物。然后加熱該均勻混合物,直至其至少軟化至可以擠塑。然后用例如雙螺桿擠塑機進行混合物擠塑,形成條形體。然后用業內已知方法將該擠出物冷卻,并切割成多顆粒。然后將多顆粒分成單位劑量。擠出物的直徑以0.1-5mm為宜,且應實現活性藥物在約8-24小時內的控釋。制備本發明熔體擠塑制劑的一種可選方法包括直接計量疏水性控釋材料,活性藥物和可選的粘合劑,加入擠塑機;加熱該均勻混合物;將均勻混合物擠塑成條;冷卻該含有均勻混合物的條形體;將其切割成大小約0.1-12mm的顆粒;將所述顆粒分成單位劑量。在這部分內容中,可實現相對連續的生產過程。可在熔體擠塑型基質中加入前文所述的增塑劑。所述增塑劑以占基質0.1-30wt%為宜。本發明控釋基質中還可包含滑石、單糖或多糖、色素、香精、潤滑劑等其他藥物賦形劑。它們的含量取決于需要獲得的特性。可通過調節擠塑機擠出孔的直徑來改變擠出條形體的厚度。而且,擠塑機的擠出口不一定要是圓形而可以是橢圓、矩形等。擠出的條形體可用熱線切割機或閘機切成顆粒。熔體擠塑型多顆粒系統的形式可以是丸粒、球粒或顆粒,這取決于擠塑機的擠出孔。就本發明目的而言,“熔體擠塑型多顆粒”和“熔體擠塑型多顆粒系統”和“熔體擠塑型顆粒”都指單位個體的集合,最好它們都具有相近的大小和/或形狀,并含有一種或多種活性藥物以及一種或多種賦形劑,并最好包含疏水性控釋材料。較好的是,所述熔體擠塑型多顆粒中顆粒的長度約為0.1-12mm,直徑約為0.1-5mm。此外,需要明白的是,所述熔體擠塑型多顆粒可以是以上大小的各種幾何形狀,例如珠、籽、丸粒等。或者,可以不經球化過程而直接將擠出物切割成所需長度的活性藥物單位劑型。在一優選實施方式中,所制備的口服劑型將有效量的熔體擠塑型多顆粒裝入膠囊。例如,可將大量熔體擠塑型多顆粒裝入明膠膠囊,裝量應足以在被攝食和接觸胃液時提供有效控釋劑量。另一優選實施方式中,適量的多顆粒擠出物經標準常規壓片技術壓成口服片。制造片劑(壓制片或模制片)、膠囊(硬、軟明膠膠囊)和丸劑的技術和配方還可參見remington制藥學(編輯:arthurosol),1553-1593(1980)。另一優選實施方式中,可如美國專利4,957,681(klimesch等)所述將擠出物成形為片劑。可選的是,控釋基質多顆粒系統或片劑或明膠膠囊可外裹以前文所述的控釋包衣。所述包衣宜包含足量疏水性和/或親水性控釋材料,以使得重量增加約2-25wt%,但是,所述包衣在更大程度上取決于具體所用阿片類鎮痛劑的物理特性和所需的釋放速度等因素。本發明劑型還可包括含一種或多種阿片類鎮痛劑的熔體擠塑型多顆粒的混合物。而且,所述劑型還可包括一定量的速釋藥物以獲得即時療效。所述速釋藥物可以是包含在明膠膠囊內的其他丸粒,或者包裹在珠粒或熔體擠塑型多顆粒之外。為獲得所需效果,本發明的單位劑型還可包含例如控釋珠粒和基質多顆粒的混合物。較好的是,本發明控釋劑型可在被攝食并先后接觸胃液和腸液時緩慢釋放出治療活性藥物。本發明熔體擠塑型制劑的控釋曲線可以通過例如改變控釋材料的量,改變增塑劑含量相對其他基質組份、疏水性材料的比例,加入其他成分或賦形劑,改變制造方法等加以改變。在本發明其他實施方式中,制備的熔體擠塑型制劑不含治療活性藥物,藥物在擠塑后加入擠出物。這樣的制劑一般是將治療活性藥物與擠出的基質材料混合,然后壓片成緩釋劑型。這樣的制劑適合治療活性藥物對軟化疏水材料和/或緩釋材料所需溫度敏感的情形。適合本發明的典型的熔體擠塑生產系統包括:合適的擠塑機驅動馬達,其速度可調但扭距恒定,具有開-關控制和電流計。此外,該生產系統應具有溫控平臺,其中包括沿擠塑機全長安裝的溫度傳感器,冷卻機構和溫度顯示機構。此外,該生產系統應包括諸如雙螺桿擠塑機之類擠塑機,所述雙螺桿擠塑機具有兩根反向旋轉的相互嚙合的螺桿,它們位于所在筒體內,該筒體的出口處有一孔或模頭。物料由加料斗進入,由螺桿推動沿筒體移動,受壓通過模頭后形成條形體,然后由例如運輸帶運去冷卻,并運至制丸機或其他合適的機械,將擠出的條形體制成多顆粒系統。制丸機可由輥、固定刀片、旋轉切割機等構成。合適的設備和系統可購自c.w.brabenderinstruments,inc.,southhackensack,newjersey等。其他合適的設備是本領域一般技術人員所公知的。本發明還涉及熔體擠塑型多顆粒的制備方法,該方法需控制擠出物中的空氣含量。通過控制擠出物中的空氣含量,我們驚奇地發現,治療活性藥物從例如多顆粒擠出物中釋放的速度被明顯改變。部分實施例中,我們驚奇地發現,擠出產物的ph依賴性也被改變。因此,在本發明的另一部分內容中,在制備熔體擠塑產物的擠塑期間需將空氣基本排除。這可以通過例如用附帶真空機構的leistritz擠塑機實現。出人意料的是,本發明用leistritz擠塑機在真空下制備的擠塑型多顆粒具有顯著不同的物理特性。具體地說,用例如提供掃描電子顯微照片(sem)的掃描電子顯微鏡觀察,該擠出物基本上沒有孔隙。雖然這與一般想像相反,但我們發現,這種基本無孔隙制劑釋放治療活性藥物的速度比非真空制備制劑快。真空下擠塑多顆粒的sem看上去十分光滑,多顆粒的牢度高于非真空制備的多顆粒。已發現,至少在部分制劑中,真空擠塑形成的擠塑型多顆粒ph依賴性比非真空下制備的同等制劑高。制備基質珠的方法本發明的控釋劑型可制備成基質珠制劑的形式。所述基質珠包含球化劑和氫可酮。氫可酮宜占基質珠重量的約0.01-99wt%,以0.1-50wt%為佳。可用來制備本發明基質珠的球化劑包括各種已知球化劑,優選纖維素衍生物,尤其是微晶纖維素。合適的微晶纖維素是例如avicelph101(商標,fmccorporation)。球化劑宜占基質珠重量的約1-99wt%。除活性成分和球化劑之外,球狀物中還可含有粘合劑。合適的粘合劑,例如低粘度水溶性聚合物類,是制藥業熟練技術人員所熟知的。然而,優選的是水溶性羥基低級烷基纖維素,例如羥丙基纖維素。除阿片類鎮痛劑和球化劑之外,本發明基質珠制劑還包含前述控釋材料,優選的有丙烯酸及甲基丙烯酸聚合物或共聚物,以及乙基纖維素。包含于本發明制劑中時,所述控釋材料的含量為基質珠的約1-80wt%。較好的是,所述控釋材料在基質珠制劑中的含量足以實現阿片類鎮痛劑從珠體中的有控釋放。基質珠制劑中還可以加有粘合劑、稀釋劑等藥物加工助劑。它們在助劑中的含量取決于需要制劑表現出的特性。可在所述基質珠之外包裹一層含有前述控釋材料的控釋包衣。該控釋包衣使得原基質珠增重約5-30%。控釋包衣的量取決于許多因素,例如基質珠的組成,以及阿片類鎮痛劑(即氫可酮)的化學和/或物理特性。基質珠一般通過將球化劑與阿片類鎮痛劑混合在一起進行例如濕法造粒來制備。然后,將所得顆粒球化成基質珠。然后,任選地用前述方法在基質珠外包裹以控釋包衣。制備基質珠的另一種方法是,例如:(a)制備含至少一種水溶性羥烷基纖維素和阿片樣物質或其鹽的顆粒;(b)將含有羥烷基纖維素的顆粒與至少一種c12-36脂肪醇混合;和(c)對顆粒進行壓片和成形,該步驟可選。較好的是,顆粒通過用水對羥烷基纖維素/阿片樣物質進行濕法造粒制得。在該方法特別優選的一種實施方式中,濕法造粒步驟中的加水量為阿片樣物質重量的1.5-5倍,1.75-3.5倍更好。另一實施方式中,可將球化劑與活性成分一起球化。所述球化劑優選微晶纖維素。合適的微晶纖維素是例如avicelph101(商標,fmccorporation)。此類實施方式中,除活性成分和球化劑之外,球粒中還可包含粘合劑。合適的粘合劑,例如低粘度水溶性聚合物,是制藥業技術人員所熟知的。然而,優選的是水溶性羥基低級烷基纖維素,例如羥丙基纖維素。此外(或者),球粒可包含一種水不溶性聚合物,尤其是丙烯酸類聚合物,丙烯酸類共聚物,例如甲基丙烯酸-丙烯酸乙酯共聚物,或乙基纖維素。此類實施方式中,緩釋包衣一般包含以下水不溶性材料,例如(a)單獨的蠟或它們與脂肪醇的混合物;或(b)蟲漆或玉米蛋白。控釋珠制劑在一特別優選的實施方式中,所述口服劑型是包含在明膠膠囊內的有效量的控釋球粒。在本發明另一優選實施方式中,所述控釋劑型是裹有含控釋材料的控釋包衣的含活性成分的球粒。“球粒”在制藥業指直徑約0.1-2.5mm,尤其是0.5-2mm之間的球形顆粒。較好的是,這些球粒外面裹有控釋材料包衣膜,該包衣可在水性介質中有控地釋放出阿片樣物質或其鹽。該包衣膜的選擇以能獲得前文所述體外釋放速度(例如1小時后釋放率至少約12.5%)等特性為宜。本發明的控釋包衣配方應能形成光滑、美觀、能承載色素等其他包衣添加劑、無毒、惰性且無粘性的牢固而連續的膜。包衣本發明制劑可以任選地裹以一種或多種適合調節釋放或保護制劑的包衣。實施方式之一中,包衣的目的是為了實現在例如與胃腸液接觸時的ph依賴性或非ph依賴性釋放。如果需要非ph依賴性釋放,設計的包衣應在周圍液體(例如胃腸道液體)中不論ph如何變化始終保持最佳釋放狀態。另一種優選實施方式是ph依賴性包衣,它可以在胃腸道的所需部位,例如胃或小腸,釋放阿片樣物質,這樣得到的吸收曲線能夠在至少12小時,更好的的24小時為患者提供鎮痛作用。還可以配制成在胃腸道一處,例如胃部,釋放一部分劑量,然后在另一處,例如小腸,釋放其余部分。采用ph依賴性包衣的本發明制劑還可以產生重復作用效應,因此,將無保護藥物包裹在腸溶包衣之外以在胃內釋放,被腸溶包衣所保護的其余部分則在以后的消化道內釋放。可用于本發明的ph依賴性包衣含有諸如蟲漆、纖維素乙酸鄰苯二甲酸酯(cap)、聚乙烯乙酸鄰苯二甲酸酯(pvap)、羥丙基甲基纖維素鄰苯二甲酸酯、甲基丙烯酸酯共聚物、玉米蛋白等控釋材料。本發明另一優選實施方式是一種穩定化的固體控釋劑型,其中含有裹有疏水性控釋材料的阿片樣物質,所需控釋材料選自:(i)烷基纖維素;(ii)丙烯酸類聚合物;(iii)它們的混合物。所述包衣可由有機或水性溶液或分散系形成。部分優選實施方式中,所述控釋包衣由疏水性控釋材料的水性分散系形成。然后將含有阿片樣物質的被包裹基質(例如片芯或惰性的藥物珠粒或球粒)固化至基質具有穩定的溶出特性。該固化終點的確定可通過將剛固化后劑型的溶出曲線與經受加速保存條件作用后(例如,40℃,75%相對濕度作用至少一個月)劑型的溶出曲線比較。此類制劑可參見美國專利5,273,760和5,286,493,它們均已轉讓于本發明的受讓人。可用于本發明的其他控釋制劑和包衣還包括美國專利5,324,351,5,356,467和5,472,712所述。在優選實施方式中,所述控釋包衣含有增塑劑。部分實施方式中,有必要在含有阿片類鎮痛劑的基質外包裹以足量的含有烷基纖維素或丙烯酸類聚合物的水分散系,以增加約2-50wt%(例如約2-25%)的重量,由此獲得控釋制劑。這層外包衣或多或少地取決于治療活性藥物的物理特性,要求的釋放率,水性分散系中是否包含增塑劑及其混合的方式等因素。烷基纖維素聚合物包括烷基纖維素在內的纖維素類材料和聚合物是適合在本發明中包裹珠粒、片劑等基質的控釋材料。例如,優選的烷基纖維素聚合物之一是乙基纖維素,但本領域技術人員知道,其他纖維素和/或烷基纖維素聚合物也可以單獨或混合用作本發明疏水性包衣的全部或一部分。一種市售的乙基纖維素水分散系是(fmccorp.,philadelphia,pennsylvania,usa)。的制備為:先將乙基纖維素溶于非水混溶性有機溶劑中,然后在表面活性劑和穩定劑存在下將其乳化于水中。均化成亞微液滴后,真空蒸發有機溶劑,形成假膠乳。在制造過程中,假膠乳中不加增塑劑。因此,在用作包衣之前,必需先將與合適的增塑劑充分混合。另一種市售乙基纖維素水分散系是(colorcon,inc.,westpoint,pennsylvania,usa)。該產品在其制造過程中即將增塑劑加入了分散系。先將聚合物熱熔體、增塑劑(癸二酸二丁酯)和穩定劑(油酸)制備成均勻的混合物,然后用堿性溶液稀釋,得到可直接加到基質上的水分散系。丙烯酸類聚合物在本發明的另一些實施方式中,含控釋材料的控釋包衣是藥學上認可的丙烯酸類聚合物,這包括但不限于:丙烯酸和甲基丙烯酸共聚物,甲基丙烯酸甲酯共聚物,甲基丙烯酸乙氧基乙酯,甲基丙烯酸氰基乙酯,聚(丙烯酸),聚(甲基丙烯酸酯),甲基丙烯酸烷酰胺共聚物,聚(甲基丙烯酸甲酯),聚甲基丙烯酸酯。聚(甲基丙烯酸甲酯)共聚物,聚丙烯酰胺,甲基丙烯酸氨基烷酯共聚物,聚(甲基丙烯酸酐)和甲基丙烯酸縮水甘油酯共聚物。部分實施方式中,所述丙烯酸類聚合物包含一種或多種銨甲基丙烯酸酯共聚物。銨甲基丙烯酸酯共聚物是業內熟知的,可參見nfxvii所述的含少量季銨基團的丙烯酸酯和甲基丙烯酸酯的全聚合共聚物。為了獲得所需的溶出曲線,或許有必要加入兩種或更多種物理特性不同的銨甲基丙烯酸酯共聚物,所述不同特性是例如不同的季銨基團與中性(甲基)丙烯酸酯的摩爾比。有些甲基丙烯酸酯類聚合物可用于制備本發明的ph依賴性包衣。例如,有一族共聚物,由甲基丙烯酸二乙基氨基乙酯與其他中性甲基丙烯酸酯合成,又稱甲基丙烯酸共聚物或聚甲基丙烯酸酯,市售的有rohmtech,inc的有數種不同類型。例如,eudragite是一種會在酸性介質中溶脹并溶出的甲基丙烯酸共聚物。eudragitl是一種在ph5.7以下不溶脹,在ph6以上溶解的甲基丙烯酸共聚物。eudragits是一種在ph6.5以下不溶脹,在ph7以上溶解的甲基丙烯酸共聚物。eudragitrl和eudragitrs在水中可溶脹,吸收的水量取決于ph,然而,用eudragitrl和eudragitrs包衣的劑型卻是非ph依賴性的。部分優選實施方式中,丙烯酸類包衣包含rohmpharma的rl30d與rs30d這兩種丙烯酸樹脂的混合物。rl30d和rs30d是低季銨含量的丙烯酸酯和甲基丙烯酸酯共聚物,其中季銨基團與其余中性(甲基)丙烯酸酯的摩爾比在rl30d中是1:20,在rs30d中是1:40。平均分子量約為150,000。rl(高滲透性)和rs(低滲透性)表示滲透性。rl/rs混合物不溶于水和消化液。然而,由它們形成的包衣可在水溶液和消化液中溶脹并被滲透。可將本發明的rl/rs分散系按任意要求比例均勻混合以制得具有所需溶出曲線的控釋制劑。例如,可由以下組成制得所需的控釋制劑:100%rl,50%rl:50%rs,以及10%rl:90%rs。當然,本領域熟練技術人員知道,也可采用其他丙烯酸類聚合物,例如l。增塑劑在包衣含有疏水性控釋材料水性分散系的實施方式中,在所述水分散系中加入有效量的增塑劑將進一步改善控釋包衣的物理特性。例如,由于乙基纖維素具有較高的玻璃轉化溫度而且在一般包衣條件下不能形成韌性膜,所以,宜在含乙基纖維素的包衣劑中加入增塑劑后用作包衣材料。通常,增塑劑在包衣溶液中的含量取決于成膜劑的濃度,例如,通常為成膜劑的約1-50wt%。然而,增塑劑的濃度只有在用具體的包衣溶液和包衣方法進行試驗后才能準確確定。適用于乙基纖維素的合適增塑劑例子包括水不溶性增塑劑,例如癸二酸二丁酯,鄰苯二甲酸二乙酯,檸檬酸三乙酯,檸檬酸三丁酯和甘油三乙酸酯,但也可以采用其他水不溶性增塑劑(例如乙酰化甘油單酯,鄰苯二甲酸酯,蓖麻油等)。就本發明乙基纖維素水分散系而言,檸檬酸三乙酯是特別好的增塑劑。適用于本發明丙烯酸類聚合物的增塑劑例子包括但不限于:檸檬酸三乙酯nfxvi,檸檬酸三丁酯等檸檬酸酯,鄰苯二甲酸二丁酯和1,2-丙二醇。還有其他增塑劑被證明可提高rl/rs樹脂溶液等所成丙烯酸類膜的彈性,這包括聚乙二醇,丙二醇,鄰苯二甲酸二乙酯,蓖麻油和甘油三乙酸酯。就本發明乙基纖維素水分散系而言,檸檬酸三乙酯是特別好的增塑劑。還發現,在控釋包衣中加入少量滑石可降低水分散系在加工過程中黏結的傾向,并可作為拋光劑。包衣珠粒制劑的制備在用疏水性材料水分散系包裹例如惰性藥物珠粒(例如nupariel18/20珠)等基質后,可將一定量的所得穩定化固體控釋珠粒裝入明膠膠囊,裝量應足以在被攝食并接觸胃腸液或溶出介質等周圍液體時給出有效的控釋劑量。本發明的穩定化控釋珠粒制劑在例如被攝食并先后接觸胃液及腸液后,可緩慢地釋放出阿片類鎮痛劑。本發明制劑的控釋曲線可通過改變疏水性控釋材料水分散系包衣量,改變增塑劑加入疏水性控釋材料水分散系的方式,改變增塑劑量相對于疏水性控釋材料的比例,加入其他成分或賦形劑,改變制造方法等加以改變。終產物的溶出曲線還可以通過例如提高或降低控釋包衣厚度加以改變。裹有治療活性藥物的基質的制備可以是例如:將治療活性藥物溶于水,然后,用wuster插管(insert)噴涂到基質(例如nupariel18/20珠)上。可選的是,還可以在包裹珠粒前加入其他成分以促進阿片樣物質與珠粒的粘合,或使溶液具有顏色等。例如,可在溶液中加入包含羥丙基甲基纖維素等并含有或不含有色素(例如colorcon,inc的產品)的產品,混合(例如約1小時)后加到基質上。然后可以在所得有包衣基質外再裹以隔離劑,以將治療活性藥物與疏水性控釋包衣隔開。合適的隔離劑之一含有羥丙基甲基纖維素。然而,各種本領域的已知成膜劑都可使用。較好的是,隔離劑不會影響終產物的溶出速度。然后可在基質外再裹以疏水性控釋材料的水分散系。疏水性控釋材料水分散系最好還包含有效量的增塑劑,例如檸檬酸三乙酯。也可使用或等預制乙基纖維素水分散系。如果采用則不必另外添加增塑劑。或者,也可采用之類預制丙烯酸類聚合物水分散系。較好的是,本發明包衣溶液除成膜劑、增塑劑和溶劑系統(即水)之外還包含色素以令產品美觀并易于區分。色素可不加入疏水性材料水分散系而加入治療活性藥物溶液中,也可以兩者都加。例如,可利用醇或丙二醇配制的色素分散系、研磨鋁色淀和二氧化鈦之類不透明劑,用剪切力將色素加入水溶性聚合物溶液,然后用低剪切力加入增塑后的由此將色素加入也可采用各種合適的方法將色素加入本發明制劑。在采用丙烯酸類聚合物時,使制劑具有顏色的合適成分包括二氧化鈦和顏料,例如氧化鐵顏料。然而,顏料的加入可能提高包衣的阻滯效應。可用各種合適的已知噴涂設備將增塑后的疏水性控釋材料水分散系噴涂到含有治療活性藥物的基質上。優選方法之一采用了wurster流化床系統,在其中,從下面注入的氣流將芯材流化,并在噴涂丙烯酸類聚合物包衣的同時進行干燥。較好的是,所涂疏水性材料水分散系的量應足以令其在接觸胃液等水性溶液時能夠實現所述治療活性藥物的預定控釋,同時應考慮到治療活性藥物的物理特性,增塑劑的加入方式等。在裹以疏水性控釋材料后,還可以再加一層成膜劑,例如加該包衣層的目的是基本上消除珠粒的聚集。還可以通過加入一種或多種釋放改變劑或提供一條或多條穿過包衣的通路來影響治療活性藥物從本發明控釋制劑中的釋放,例如將其調節至所需的釋放速度。疏水性控釋材料與水溶性材料之比取決于所需的釋放速度和所選材料的溶解度等因素。所述釋放改變劑具有致孔作用,可以是有機或無機物質,包括那些可在使用環境中從包衣內溶出、萃出或滲出的物質。該致孔劑可包含一種或多種親水性材料,例如羥丙基甲基纖維素。本發明控釋包衣還可以包含淀粉和樹膠等促腐蝕劑。本發明控釋包衣還可以包含可在使用環境中形成微孔層的材料,例如聚合物鏈上重復出現有碳酸酯基團的線性碳酸聚酯等聚碳酸酯。釋放改變劑還可以包含半滲透性聚合物。部分優選實施方式中的釋放改變劑選自羥丙基甲基纖維素,乳糖,硬脂酸金屬鹽,以及它們的混合物。本發明控釋包衣還可包含一種由至少一條通路,一個微孔等構成的滲出機制。所述通路可用以下美國專利所述方法形成:美國專利3,845,770,3,916,889,4,063,064,和4,088,864。所述通路可以是各種形狀的,例如圓形,三角形,正方形,橢圓,不規則形等。生產24小時控釋珠粒制劑的另一種方法是粉末敷層。已轉讓于本發明受讓人的美國專利5,411,745用主要由極細水合乳糖構成的加工助劑,通過粉末敷層技術制備24小時嗎啡制劑的方法。粉末敷層珠粒的制備為:將粘合劑水溶液噴涂在惰性珠粒上,形成一層粘性表面,然后向該粘性珠粒上噴涂硫酸嗎啡和極細水合乳糖混合物粉末。然后將珠粒干燥,并裹以前文所述疏水性材料,以實現最終產品在周圍液體中按要求釋放藥物。然后,將適量控釋珠粒裝入膠囊,成為可在約12小時內維持嗎啡有效血漿濃度的成品劑型。優選實施方式的詳細描述以下實施例說明了本發明的各方面內容。它們僅用以說明本發明,并不限定本發明的范圍。實施例1按照表i配方制備氫可酮緩釋片劑:表i成分單位含量(mg)批含量(g)二酒石酸氫可酮15.0150.0噴霧干燥的乳糖56.0560.0聚乙烯吡咯烷酮4.040.0eudragitrs30d(固體)10.0100.0甘油三乙酸酯2.020.0硬脂醇20.0200.0滑石2.020.0硬脂酸鎂1.010.0總量110.01100.0制備過程:1.阻滯劑分散系:用lightnin混合機混合eudragitrs30d與甘油三乙酸酯。2.熔化硬脂醇。3.用流化床造粒機將阻滯劑分散系噴涂到二酒石酸氫可酮、噴霧干燥乳糖和聚乙烯吡咯烷酮上。4.將整批產品在不銹鋼盤上干燥15分鐘,或者干燥至恒重。5.用hobart混合機將熔融硬脂醇加入該批產物。6.在不銹鋼盤上進行30分鐘干燥蠟造粒,或至造粒溫度達35℃或以下。7.用comil研磨冷卻后的造粒產物。8.用hobart混合機加入滑石和硬脂酸鎂作為潤滑劑。9.用壓片機將造粒產物壓制成片劑。然后如下測定片劑溶出率:1.儀器:usp法i(轉籃法),100rpm。2.介質:700mlsgf,55分鐘,然后,900ml無酶sif3.取樣時間:1,2,4,8和12小時4.分析方法:高效液相色譜。溶出參數如表ii所示:表ii時間(小時)溶出百分比139.7251.5467.4886.41296.1然后在一生物利用度試驗中得到實施例1(cr)和一速釋參比標準(ir)的cmax和tmax,比較15mg氫可酮以速釋制劑(lortab7.5mg×2)和以上述cr制劑形式給予健康人受試者的差異,結果見表iii:表iii實施例2按表iv配方制備氫可酮緩釋片劑:表iv成分單位含量(mg)批含量(g)二酒石酸氫可酮15.0150.0噴霧干燥的乳糖51.0510.0聚乙烯吡咯烷酮4.040.0eudragitrs30d(固體)10.0100.0甘油三乙酸酯2.020.0硬脂醇25.0250.0滑石2.020.0硬脂酸鎂1.010.0總量110.01100.0制備過程同實施例1。按照實施例1的方法獲得溶出參數,見表v:表v時間(小時)溶出百分比136245.8460.5878.91290.4實施例3按表vi配方制備氫可酮緩釋膠囊:表vi成分單位含量(mg)批含量(g)二酒石酸氫可酮15.0320.0eudragitrspo76.01520.0eudragitrlpo4.080.0硬脂醇25.0500.0總量120.02400.0制備方法如下:1.用hobart混合機研磨混合硬脂醇,eudragitrlpo,二酒石酸氫可酮和eudragitrspo。2.擠塑造粒產物:用粉末加料機,熔體擠塑機(裝有6×1mm模頭),傳送帶,lasermike和造丸機,條件如下:粉末進料速度:40g/min;螺桿速度:185rpm;真空度:980mbar傳送帶:使得擠出物的直徑為1mm造丸機:切成1mm長的丸粒3.用16號和20號篩篩選丸粒。收集通過16號篩網而滯留在20號篩網上的物料。4.將丸粒裝入2號明膠膠囊。范圍:nlt114mg,nmt126mg。然后按照實施例1的方法獲取溶出參數。結果見表vii:表vii時間(h)溶出百分比123.9234.7451.7874.61285.2實施例4按照表viii配方制備羥考酮緩釋片:表viii成分單位含量(mg)批含量(g)鹽酸羥考酮20.022.0噴霧干燥乳糖59.2565.175聚乙烯吡咯烷酮5.05.5eudragitrs30d(固體)10.011.0甘油三乙酸酯2.02.2硬脂醇25.027.5滑石2.52.75硬脂酸鎂1.251.375opadry粉紅y-s-14518a4.04.26總量129.0141.76方法如下:1.造粒:用流化床造粒機將eudragit/甘油三乙酸酯分散系噴涂在鹽酸羥考酮、噴霧干燥乳糖和聚乙烯吡咯烷酮上。2.研磨:放出造粒產物,令其通過磨機。3.涂蠟:熔化硬脂醇,用混合機加到研磨后的顆粒中。任其冷卻。4.研磨:讓冷卻后的顆粒通過磨機。5.潤滑:用混合機,在顆粒中加入滑石和硬脂酸鎂作為潤滑劑。6.壓片:用壓片機將顆粒壓制成片。7.包衣:在片劑外包裹一層水性膜。然后,如下對片劑進行溶出試驗:1.儀器:uspii型(槳法),150rpm2.介質:第一小時為700mlsgf,然后加入磷酸鹽緩沖液至900ml,ph7.5。3.取樣時間:1,2,4,8,12,18和24小時4.分析方法:高效液相色譜溶出參數見表ix:表vii時間(h)溶出百分比14525547088712961810124102然后,通過生物利用度研究獲取實施例4和標準速釋參比樣品的cmax和tmax,見表x:表x實施例5按表xi配方制備嗎啡緩釋片:表xi成分單位含量(mg)批含量(g)硫酸嗎啡30.0138.0噴霧干燥的乳糖70.0322.0羥乙基纖維素10.046.0鯨蠟硬脂醇35.0161.0滑石3.013.8硬脂酸鎂2.09.2opadryys-1-47295.023.0總量155.0713.0方法如下:1.造粒:在一混合機中,將水加入硫酸嗎啡、噴霧干燥乳糖和羥乙基纖維素,用流化床造粒機干燥。2.篩選:放出顆粒,過篩。3.涂蠟:熔化鯨蠟硬脂醇,用混合機將其加入經研磨的顆粒。任其冷卻。4.篩選:將冷卻后的顆粒過篩。5.潤滑:用混合機將滑石和硬脂酸鎂加入顆粒作為潤滑劑。6.壓片:用壓片機將顆粒壓制成片。7.包衣:在片劑外包以水性膜。如下進行片劑的溶出試驗:1.儀器:usp方法i(轉籃法),50rpm2.介質:900ml純水,37℃3.取樣時間:1,2,3,4和6小時。4.分析方法:285nm和305nmuv檢測,用5cm池,2點法。溶出參數見表xii:表xii時間(h)溶出百分比134.2249.9364.2475.5690.3然后,通過生物利用度研究測定實施例5和一標準速釋參比樣品的cmax和tmax:表xiii實施例6比較實施例1,4和5的藥物動力學參數,出人意料的發現,盡管實施例1鹽酸氫可酮控釋片的溶出非常類似于實施例4的控釋羥考酮片和實施例5的硫酸嗎啡控釋片,但氫可酮制劑cr與ir的cmax之比為38%,而羥考酮和嗎啡片劑則高于50%。比較結果見表xiv:表xiv實施例7以實施例1,2,3的控釋氫可酮制劑和兩片速釋(二酒石酸氫可酮7.5mg/對乙酰氨基酚500mg)片劑對空腹正常態志愿者進行單劑量、4次治療、標簽公開(openlable)的藥物動力學比較。各制劑的血漿濃度見表15-18:藥物動力學參數見表19:表19a:auc和cmax的幾何平均值(0,最后),tmax,w50,t1/2(abs)(吸收半衰期)和t1/2(elim)(消除半衰期)的算術平均值。b:比例和90%ci所基于的是最小二乘方平均值。c:比例(%):(試驗平均值/參比平均值)×100,所基于的是最小二乘方平均值。實施例8按表xx配方制備氫可酮緩釋片:表xx成分mg/片kg/批二酒石酸氫可酮1515.0磷酸氫鈣3131.0甘油山萮酸酯1010.0硬脂醇2222.0微晶纖維素3131.0硬脂酸鎂1.01.0opadry紫ys-1-10371-a5.05.0純水n/a128.331115.0mg115.0kg1:在加工過程中被蒸發,不包含在最終產品中方法如下:1.研磨:令硬脂醇薄片通過磨機。2.混合:用合適的混合機,將二酒石酸氫可酮、磷酸氫鈣、甘油山萮酸酯,硬脂醇和微晶纖維素混合。3.擠塑:連續地將混合物料加入雙螺桿擠塑機,擠塑機溫度被升高以軟化并形成擠出物。4.冷卻:擠出物在傳送帶上冷卻。5.研磨:將冷卻后的擠出物通過磨機,得到所需大小的顆粒。6.混合:將研磨后的擠出物與硬脂酸鎂混合。7.壓片:用壓片機將所得顆粒壓制成片。8.包衣:將opadry分散在純水中制備成包衣膜溶液,將其涂在片劑上。如下進行片劑的試驗:1.儀器:uspi型(轉籃法),100rpm。2.介質:最初55分鐘為700mlsgf(不含酶),加入磷酸鹽緩沖液至900ml,ph7.5。3.取樣時間:1,2,4,8和12小時。4.分析方法:高效液相色譜。溶出參數見表xxi:表xxi時間(h)溶出百分比1222374588841299實施例9進行如下三路交叉藥物動力學比較:給予進食和空腹常態志愿者單劑量15mg氫可酮控釋片(實施例8);給予空腹常態志愿者15mg氫可酮速釋劑(2×7.5mg/片),q6h,2次以上。然后,通過生物利用度研究獲得了實施例8和標準速釋參比片的cmax和tmax,見表xxii和xxiii:表xxii表xxiii當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1