一種酶促小分子自組裝制備納米凝膠的方法與流程
本發明屬于納米生物材料技術領域,涉及一種酶促小分子自組裝制備納米凝膠的方法。
背景技術:
水凝膠是以水為分散介質的凝膠,在具有網狀交聯結構的水溶性高分子中引入一部分疏水基團和親水殘基,親水殘基與水分子結合,將水分子連接在網狀內部,而疏水殘基遇水膨脹。
隨著水凝膠技術的不斷發展,水凝膠的研究逐步從大塊凝膠,到微凝膠,再到納米凝膠。納米凝膠是一類通過物理或者化學交聯的具有三維網絡結構的納米尺度的水凝膠微粒,通常由高分子單體聚合而成,直徑在200nm以下。納米凝膠相對于其它凝膠來說,具有以下優勢:一、尺寸小,容易被細胞吞噬;二、容易穿透人體中的各種保護膜,如腦膜,從而可以實現腦部給藥;三、載藥效率高。因此,近年來納米凝膠受到了人們越來越多的關注。
目前,納米凝膠的制備方法主要為共混法,即將無機納米粒子與聚合物按照不同的工藝復合而成,是目前制備聚合物納米復合水凝膠最簡單的一種方法,該方法的優點是納米粒子與聚合物的制備分開進行,可有效控制納米粒子的形態和尺寸,但缺點是共混時納米粒子易發生團聚,不易分散。
酶催化聚合反應是近年來研究的熱點之一,在制備水凝膠方面,由于酶具有高效、專一、穩定的特點,使得它成為了當前制備水凝膠最有發展前景的方法之一。酶催化自由基聚合屬于酶催化聚合的一種,近幾年用這種方法制備水凝膠逐漸走進了大家的視野,這種方法制得的水凝膠在調節物理和化學性質方面非常靈活,并且具有很好的酶響應性。但目前尚未有簡單易行的方法,利用酶催化聚合反應制備出性能優異的納米凝膠。
技術實現要素:
本發明的目的就是為了克服上述現有技術存在的缺陷而提供一種納米凝膠分散性好且簡單易行的酶促小分子自組裝制備納米凝膠的方法。
本發明的目的可以通過以下技術方案來實現:
一種酶促小分子自組裝制備納米凝膠的方法,該方法是先依次對納米顆粒的表面進行氨基化修飾、羧基化修飾,之后通過酰胺反應將酶修飾在納米顆粒表面,最后加入小分子肽,在酶的作用下,小分子肽轉化為凝膠因子,并在納米顆粒表面自組裝形成納米凝膠。新形成的凝膠因子優先在納米顆粒表面組裝,從而形成凝膠包裹在納米顆粒表面。
該方法具體包括以下步驟:
(1)納米顆粒的氨基化修飾:將納米顆粒分散在溶劑中,之后加入氨基硅烷偶聯劑,并在40-70℃下反應8-24h,洗滌后得到氨基化修飾的納米顆粒;
(2)納米顆粒的羧基化修飾:將氨基化修飾的納米顆粒分散在有機溶劑中,之后加入丁二酸酐,并攪拌6-18h,洗滌后得到羧基化修飾的納米顆粒;
(3)納米顆粒的酶修飾:將羧基化修飾的納米顆粒分散在生物緩沖液中,并調節ph值至5-6,活化羧基后,加入酶并進行酰胺反應,洗滌后得到酶修飾的納米顆粒;
(4)納米顆粒表面小分子自組裝制備納米凝膠:將酶修飾的納米顆粒分散在溶劑中,并調節ph值至7-8,之后加入小分子肽,攪拌反應6-12h后,即得到所述的納米凝膠。
所述的納米顆粒包括二氧化硅納米顆粒或四氧化三鐵納米顆粒中的一種或兩種。其中,二氧化硅納米顆粒采用
所述的納米顆粒的粒徑為180-220nm。優選為200nm,200nm的尺寸有利于后續的生物應用,例如比較容易穿透屏障到達腫瘤癌癥等病變部位,用于藥物載體,由于被動運輸的作用,能夠實現藥物控釋和靶向治療。
步驟(1)及步驟(4)中,所述的溶劑包括水或乙醇中的一種或兩種;步驟(2)中,所述的有機溶劑包括n,n-二甲基甲酰胺或n-甲基吡咯烷酮中的一種或兩種;步驟(3)中,所述的生物緩沖液包括2-嗎啉乙磺酸或檸檬酸中的一種或兩種。
步驟(1)中,所述的氨基硅烷偶聯劑包括3-氨丙基三乙氧基硅烷或3-氨丙基三甲氧基硅烷中的一種或兩種;步驟(3)中,所述的酶包括羧酸酯酶或酸性磷酸酶中的一種或兩種。
步驟(4)中,所述的小分子肽包括napffes或fmoc-tyr(h2po3)-oh中的一種或兩種,所述的napffes的化學結構式如下式所示:
所述的fmoc-tyr(h2po3)-oh的化學結構式如下式所示:
步驟(3)中,所述的活化羧基過程為:加入nhs及edc,之后攪拌1.5-2.5h即可,每100ml生物緩沖液中分別加入100-120mg所述的nhs及220-240mg所述的edc。nhs為n-羥基琥珀酰亞胺,edc為1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽。
步驟(3)中所述的酰胺反應過程中,反應溫度為15-35℃,反應時間為5-7h。
步驟(1)中,每100ml溶劑中,分別加入800-1200mg所述的納米顆粒及0.8-0.9ml所述的氨基硅烷偶聯劑;
步驟(2)中,每100ml有機溶劑中,分別加入800-1200mg所述的氨基化修飾的納米顆粒及80-120mg所述的丁二酸酐;
步驟(3)中,每100ml生物緩沖液中,分別加入80-120mg所述的羧基化修飾的納米顆粒及40-60mg所述的酶;
步驟(4)中,每100ml溶劑中,分別加入80-120mg所述的酶修飾的納米顆粒及15-25mg所述的小分子肽。
與現有技術相比,本發明具有以下特點:
1)通過酶促進小分子肽轉化為凝膠因子,并優先在納米顆粒表面組裝成小分子凝膠,進而制備出納米凝膠,該納米凝膠具有生物活性和多功能性,可作為生長因子、藥物及細胞的載體,應用于細胞培養、傷口修復的治療等,為納米凝膠的制備提供了新的思路,豐富了納米凝膠的制備方法,且本發明具有溫和高效、生物相容性強等優勢,大大減少了各類溶劑清洗的時間和成本,有利于后續的生物顯影、hifu超聲等生物應用;
2)通過改變小分子肽的濃度,能夠實現凝膠層的厚度可控,隨著凝膠層厚度的增加,表面電勢會發生相應改變,且該體系成膠安全高效,具有良好的強度和生物活性,能夠在應用在細胞培養或組織工程等領域;
3)可通過改變各組分的濃度或用量,調控納米凝膠的交聯率與成膠速度,并可根據需要實現原位成膠,靈活性好。
附圖說明
圖1為實施例1中納米凝膠的制備過程示意圖;
圖2為實施例1中制備得到的napffes的nmr圖譜;
圖3為實施例1中制備得到的sio2納米顆粒及納米凝膠的sem圖譜;
圖4為實施例2中納米凝膠的制備過程示意圖;
圖5為實施例1中制備得到的fe3o4納米顆粒及納米凝膠的sem圖譜。
具體實施方式
下面結合附圖和具體實施例對本發明進行詳細說明。本實施例以本發明技術方案為前提進行實施,給出了詳細的實施方式和具體的操作過程,但本發明的保護范圍不限于下述的實施例。
實施例1:
一種酶促小分子自組裝制備納米凝膠的方法如圖1所示,具體包括以下步驟:
(1)sio2納米顆粒的合成:
將147ml無水乙醇、2.5ml去離子水、7.5ml氨水于40℃下攪拌均勻,再向其中加入2.8ml正硅酸乙酯水解3h,之后離心,并用無水乙醇洗滌三次,得到硅球,即sio2納米顆粒。
(2)固相合成法制備napff:
(2-1)稱取0.3g(0.405mmol)2-氯三苯甲基氯樹脂于固相合成器中,并加入2.5mldmf,之后通入氮氣5min,使2-氯三苯甲基氯樹脂充分溶脹;
(2-2)用氮氣把二氯甲烷從裝有2-氯三苯甲基氯樹脂的固相合成器中壓除干凈;
(2-3)將314mg(0.81mmol)fmoc保護的氨基酸溶解在2ml的dmf里,加入145μl(0.83mmol)diea(n,n-二異丙基乙胺),然后轉移到上述固相合成器中,通入氮氣,在室溫下反應2h;
(2-4)加入130μl甲醇于固相合成器中,反應5分鐘,把該固相合成器中液體壓凈,用2.5mldmf洗滌三次,每次30s,再用5ml含哌啶體積百分比20%的dmf洗滌,最后用2.5mldmf清洗3次,每次30s;
(2-5)取第二個fmoc保護的氨基酸256mg(0.673mmol)、120μl(0.573mmol)diea和2.5ml無水dmf制成溶液,把配好的溶液加入到上述固相合成器中,通入氮氣反應2h;
(2-6)重復(2-5)方法依次加入所有需要的氨基酸,然后用dmf洗滌3遍,再用環己烷和異丙醇各洗三遍,每次1ml;
(2-7)按三氟乙酸與無水二氯甲烷的體積比1:9,配制成體積百分比濃度為10%的三氟乙酸溶液,把產物從2-氯三苯甲基氯樹脂上切下,濃縮,加入ph=3稀鹽酸以除去殘留的三氟乙酸,用油泵抽干,得到粗品,之后用hplc分離提純,即制得napff,其化學結構式如下式所示:
(3)napffes的合成:
(3-1)將480mg(1mmol)napff、115mg(1mmol)nhs溶解在20mlthf里,之后加入230mg(1.2mmol)edc,活化2h,再加入過量的乙醇胺(2ml)反應6h,然后旋蒸濃縮,用ph=3的稀鹽酸洗滌三次,凍干,得到固體;
(3-2)將(3-1)反應得到的固體溶于20ml三氯甲烷中,加入3mmol(0.5ml)diea(n,n-二異丙基乙胺)和3mmol(300mg)丁二酸酐,反應一晚上,過濾,取其濾液旋蒸,用ph=3稀鹽酸洗滌三次,凍干得到粗品,用hplc分離提純,即得到napffes。
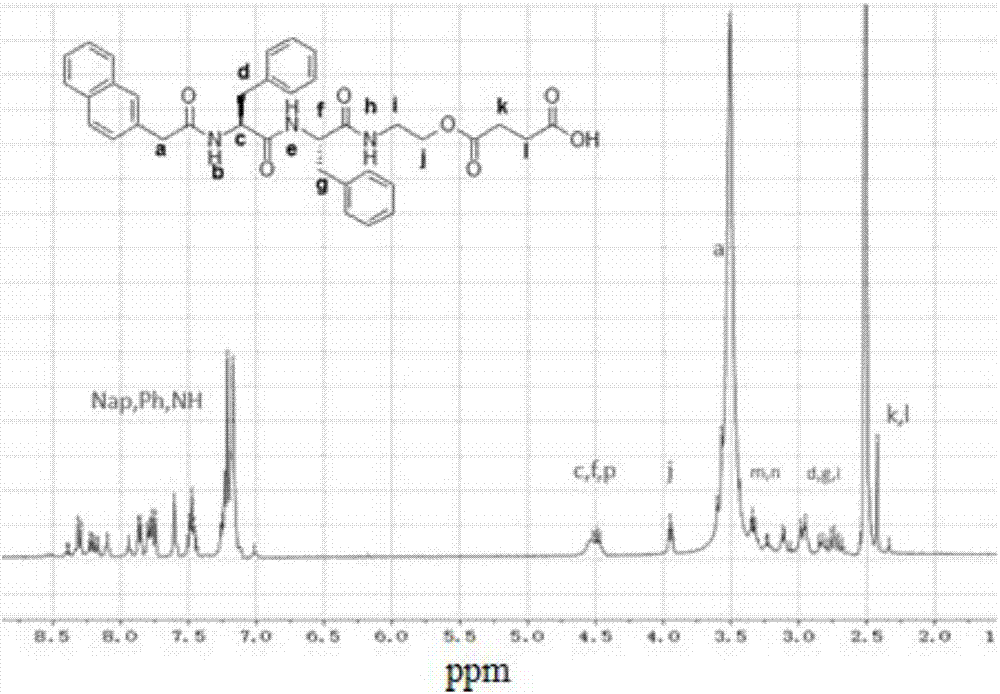
圖2為制備得到的napffes的nmr圖譜,由圖2可以看出,通過上述步驟,合成出了小分子肽napffes。
(4)硅球表面氨基化修飾:將(1)中制備好的硅球,分散到120ml乙醇中,于40℃下攪拌,并向其中加入1ml的氨基硅烷偶聯劑(3-氨丙基三乙氧基硅烷),反應一晚上,用乙醇洗滌數遍。
(5)硅球表面羧基化修飾:將(4)中制備好的氨基化的硅球用乙醇洗滌后分散到dmf中,向其中加入三倍的diea(1ml)和三倍的丁二酸酐(100mg),攪拌6h,反應完全后用乙醇洗滌三遍,再用去離子水洗滌三遍,凍干以備用。
(6)硅球表面修飾酯酶:將(5)羧基化的硅球分散到去2-嗎啉乙磺酸100ml中,調節ph,使ph約5.5左右,加入1mmolnhs(115mg)、1.2mmoledc(230mg)活化羧基2h,然后加入50mg的羧酸酯酶室溫攪拌6h,之后用去離子水洗滌三次,凍干后備用。
(7)硅球表面小分子自組裝納米凝膠制備:100mg修飾好酯酶的硅球分散到100ml去離子水中,調節ph使ph約等于7.5,向其中加入20mg的napffes反應一晚上,在酯酶作用下前驅體napffes變為凝膠因子napffe,在二氧化硅表面自組裝形成納米凝膠。然后洗滌超聲,重新分散到水里得到納米凝膠,或者凍干以備用。其中,napffes化學結構式如下式所示:
圖3為制備得到的sio2納米顆粒及納米凝膠的sem圖譜,由圖4可以看出,制備得到的納米凝膠中,sio2納米顆粒表面裹有一層水凝膠層。
實施例2:
一種酶促小分子自組裝制備納米凝膠的方法如圖4所示,具體包括以下步驟:
(1)fe3o4納米顆粒的合成:
先將0.54gfecl3溶解于20ml乙二醇和二乙二醇的混合溶液中(veg/vdeg=3/17),攪拌30min后,加入2g聚乙烯吡咯烷酮(pvp,k30),然后加熱至120℃,并在攪拌下通入n2,形成透明溶液,反應1h后,加入1.5gnaoac,并停止加熱,劇烈攪拌30min后,將溶液轉移至50ml聚四氟乙烯反應釜中,再將反應釜置于200℃烘箱中保持12h。反應完成后冷卻至室溫,取出反應釜,得到的黑色產物即磁性納米四氧化三鐵粒子(mnps),先后用乙醇和去離子水清洗幾次,之后分散在20ml去離子水中備用。
(2)四氧化三鐵(fe3o4)納米顆粒的表面修飾
取2ml上述制備的fe3o4分散在80ml乙醇和80ml去離子水的混合液中,超聲分散30min后,加入6mlaptes(3-氨丙基三乙氧基硅烷),并在70℃加熱攪拌下反應24h,同時通入n2。反應完成后,分別用乙醇和去離子水清洗幾次,得到的顆粒為表面氨基化的四氧化三鐵,即fe3o4-aptes。接著將fe3o4-aptes分散在100mldmf中,再加入10%的丁二酸酐,攪拌反應18h,之后用乙醇和水清洗,得到表面羧基化的四氧化三鐵fe3o4-cooh,將fe3o4-cooh分散在20ml去離子水中備用。
(3)四氧化三鐵(fe3o4)納米水凝膠的制備
將fe3o4-cooh用酸性磷酸酶(ap)修飾后,分散于100ml濃度為0.5mg/ml的fmoc-tyr(h2po3)-oh溶液中,超聲分散后,攪拌一定時間,待自組裝完成后磁分離除去未反應的物質,再用去離子水清洗三次得到納米凝膠,將納米凝膠分散在去離子水中備用。
四氧化三鐵納米水凝膠在制備過程中,通過酸性磷酸酶的酶切作用除去fmoc-tyr(h2po3)-oh中的磷酸根,使小分子fmoc-tyr(h2po3)-oh變為fmoc-tyr-oh,由親水性變為疏水性,fmoc-tyr-oh由于疏水性在fe3o4顆粒表面自組裝,形成一層小分子凝膠。
圖5為制備得到的fe3o4納米顆粒及納米凝膠的sem圖譜,由圖5可以看出,制備得到的納米凝膠中,fe3o4納米顆粒表面裹有一層水凝膠層。
實施例3:
一種酶促小分子自組裝制備納米凝膠的方法,該方法是先依次對納米顆粒的表面進行氨基化修飾、羧基化修飾,之后通過酰胺反應將酶修飾在納米顆粒表面,最后加入小分子肽,在酶的作用下,小分子肽轉化為凝膠因子,并在納米顆粒表面自組裝形成納米凝膠。
該方法具體包括以下步驟:
(1)納米顆粒的氨基化修飾:將納米顆粒分散在溶劑中,之后加入氨基硅烷偶聯劑,并在40℃下反應24h,洗滌后得到氨基化修飾的納米顆粒,其中,每100ml溶劑中,分別加入800mg納米顆粒及0.9ml氨基硅烷偶聯劑;
(2)納米顆粒的羧基化修飾:將氨基化修飾的納米顆粒分散在有機溶劑中,之后加入丁二酸酐,并攪拌12h,洗滌后得到羧基化修飾的納米顆粒,其中,每100ml有機溶劑中,分別加入1000mg氨基化修飾的納米顆粒及100mg丁二酸酐;
(3)納米顆粒的酶修飾:將羧基化修飾的納米顆粒分散在生物緩沖液中,并調節ph值至6,之后加入nhs及edc,并攪拌1.5h活化羧基,再加入酶,并在35℃進行酰胺反應5h,洗滌后得到酶修飾的納米顆粒,其中,每100ml生物緩沖液中,分別加入120mgnhs、220mgedc、120mg羧基化修飾的納米顆粒及40mg酶;
(4)納米顆粒表面小分子自組裝制備納米凝膠:將酶修飾的納米顆粒分散在溶劑中,并調節ph值至7.5,之后加入小分子肽,攪拌反應9h后,即得到納米凝膠,其中,每100ml溶劑中,分別加入100mg酶修飾的納米顆粒及20mg小分子肽。
其中,納米顆粒為二氧化硅納米顆粒,納米顆粒的粒徑為200nm。步驟(1)及步驟(4)中,溶劑為水;步驟(2)中,有機溶劑為n-甲基吡咯烷酮;步驟(3)中,生物緩沖液為2-嗎啉乙磺酸。步驟(1)中,氨基硅烷偶聯劑包括3-氨丙基三乙氧基硅烷和3-氨丙基三甲氧基硅烷;步驟(3)中,酶為羧酸酯酶。步驟(4)中,小分子肽為napffes,napffes的化學結構式如下式所示:
實施例4:
一種酶促小分子自組裝制備納米凝膠的方法,該方法是先依次對納米顆粒的表面進行氨基化修飾、羧基化修飾,之后通過酰胺反應將酶修飾在納米顆粒表面,最后加入小分子肽,在酶的作用下,小分子肽轉化為凝膠因子,并在納米顆粒表面自組裝形成納米凝膠。
該方法具體包括以下步驟:
(1)納米顆粒的氨基化修飾:將納米顆粒分散在溶劑中,之后加入氨基硅烷偶聯劑,并在70℃下反應8h,洗滌后得到氨基化修飾的納米顆粒,其中,每100ml溶劑中,分別加入1200mg納米顆粒及0.8ml氨基硅烷偶聯劑;
(2)納米顆粒的羧基化修飾:將氨基化修飾的納米顆粒分散在有機溶劑中,之后加入丁二酸酐,并攪拌18h,洗滌后得到羧基化修飾的納米顆粒,其中,每100ml有機溶劑中,分別加入800mg氨基化修飾的納米顆粒及120mg丁二酸酐;
(3)納米顆粒的酶修飾:將羧基化修飾的納米顆粒分散在生物緩沖液中,并調節ph值至5,之后加入nhs及edc,并攪拌2.5h活化羧基,再加入酶,并在15℃進行酰胺反應7h,洗滌后得到酶修飾的納米顆粒,其中,每100ml生物緩沖液中,分別加入100mgnhs、240mgedc、80mg羧基化修飾的納米顆粒及60mg酶;
(4)納米顆粒表面小分子自組裝制備納米凝膠:將酶修飾的納米顆粒分散在溶劑中,并調節ph值至7,之后加入小分子肽,攪拌反應12h后,即得到納米凝膠,其中,每100ml溶劑中,分別加入80mg酶修飾的納米顆粒及25mg小分子肽。
其中,納米顆粒為四氧化三鐵納米顆粒,納米顆粒的粒徑為220nm。步驟(1)及步驟(4)中,溶劑為乙醇;步驟(2)中,有機溶劑包括n,n-二甲基甲酰胺和n-甲基吡咯烷酮;步驟(3)中,生物緩沖液包括2-嗎啉乙磺酸和檸檬酸。步驟(1)中,氨基硅烷偶聯劑為3-氨丙基三甲氧基硅烷;步驟(3)中,酶為酸性磷酸酶。步驟(4)中,小分子肽為fmoc-tyr(h2po3)-oh。
實施例5:
一種酶促小分子自組裝制備納米凝膠的方法,該方法是先依次對納米顆粒的表面進行氨基化修飾、羧基化修飾,之后通過酰胺反應將酶修飾在納米顆粒表面,最后加入小分子肽,在酶的作用下,小分子肽轉化為凝膠因子,并在納米顆粒表面自組裝形成納米凝膠。
該方法具體包括以下步驟:
(1)納米顆粒的氨基化修飾:將納米顆粒分散在溶劑中,之后加入氨基硅烷偶聯劑,并在55℃下反應16h,洗滌后得到氨基化修飾的納米顆粒,其中,每100ml溶劑中,分別加入1000mg納米顆粒及0.85ml氨基硅烷偶聯劑;
(2)納米顆粒的羧基化修飾:將氨基化修飾的納米顆粒分散在有機溶劑中,之后加入丁二酸酐,并攪拌6h,洗滌后得到羧基化修飾的納米顆粒,其中,每100ml有機溶劑中,分別加入1200mg氨基化修飾的納米顆粒及80mg丁二酸酐;
(3)納米顆粒的酶修飾:將羧基化修飾的納米顆粒分散在生物緩沖液中,并調節ph值至5.5,之后加入nhs及edc,并攪拌2h活化羧基,再加入酶,并在20℃進行酰胺反應6h,洗滌后得到酶修飾的納米顆粒,其中,每100ml生物緩沖液中,分別加入110mgnhs、230mgedc、100mg羧基化修飾的納米顆粒及50mg酶;
(4)納米顆粒表面小分子自組裝制備納米凝膠:將酶修飾的納米顆粒分散在溶劑中,并調節ph值至8,之后加入小分子肽,攪拌反應6h后,即得到納米凝膠,其中,每100ml溶劑中,分別加入120mg酶修飾的納米顆粒及15mg小分子肽。
其中,納米顆粒包括二氧化硅納米顆粒和四氧化三鐵納米顆粒,納米顆粒的粒徑為180nm。步驟(1)及步驟(4)中,溶劑包括水和乙醇;步驟(2)中,有機溶劑為n,n-二甲基甲酰胺;步驟(3)中,生物緩沖液為檸檬酸。步驟(1)中,氨基硅烷偶聯劑為3-氨丙基三乙氧基硅烷;步驟(3)中,酶包括羧酸酯酶和酸性磷酸酶。步驟(4)中,小分子肽包括napffes和fmoc-tyr(h2po3)-oh,napffes的化學結構式如下式所示:
上述的對實施例的描述是為便于該技術領域的普通技術人員能理解和使用發明。熟悉本領域技術的人員顯然可以容易地對這些實施例做出各種修改,并把在此說明的一般原理應用到其他實施例中而不必經過創造性的勞動。因此,本發明不限于上述實施例,本領域技術人員根據本發明的揭示,不脫離本發明范疇所做出的改進和修改都應該在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種具有葡萄糖敏感性和溫度敏感性的自組裝納米粒子及其制備方法
- 一種基于納米材料自組裝的土霉素sers檢測方法
- 暴露高能晶面納米片自組裝的三維網絡結構α?Fe<sub>2</sub>O<sub>3</sub>的制備方法
- 一種三氧化鎢納米帶自組裝成微納米星的制備方法
- 一種葡萄糖和溫度敏感性的自組裝納米粒子及其制備方法和應用
- 允許通過嵌段共聚物的自組裝產生納米結構體的方法
- 一種自組裝納米止血劑、其制備方法及應用
- 一種納米片自組裝棱臺狀(NH<sub>4</sub>)<sub>2</sub>V<sub>3</sub>O<sub>8</sub>的制備方法
- 一種干燥介導金納米顆粒自組裝制備金基底的方法及其應用
- 一種以桿菌肽為模板自組裝合成球狀金納米粒子的方法
- 一種基于納米材料自組裝的土霉素sers檢測方法
- 暴露高能晶面納米片自組裝的三維網絡結構α?Fe<sub>2</sub>O<sub>3</sub>的制備方法
- 一種三氧化鎢納米帶自組裝成微納米星的制備方法
- 一種葡萄糖和溫度敏感性的自組裝納米粒子及其制備方法和應用
- 允許通過嵌段共聚物的自組裝產生納米結構體的方法
- 一種自組裝納米止血劑、其制備方法及應用
- 一種納米片自組裝棱臺狀(NH<sub>4</sub>)<sub>2</sub>V<sub>3</sub>O<sub>8</sub>的制備方法
- 一種干燥介導金納米顆粒自組裝制備金基底的方法及其應用
- 一種以桿菌肽為模板自組裝合成球狀金納米粒子的方法
- 一種果膠-多臂聚乙二醇自組裝制備納米藥物的方法