一種曲普瑞林微球及其制備方法與應用與流程
本發明屬于藥物制劑領域,具體涉及一種曲普瑞林微球、其制備方法以及用于制備注射劑的用途。
背景技術:
曲普瑞林是一種人工合成的十肽,其在天然的促黃體生成激素釋放激素(LHRH)的基礎上利用D-trp將LHRH肽鏈第六位上的Gly取代,得到生物活性更強的LHRH類似物(LHRHa)。曲普瑞林在臨床上主要用于治療子宮肌瘤、兒童中樞性性早熟、子宮內膜異位癥、不孕癥及男性前列腺癌等,其臨床應用與其激素生理作用密切相關。曲普瑞林的激素功效因給藥方式和劑量的不同具體體現為正向的促進和負向的抑制,以生理性脈沖給藥,對性腺軸是興奮作用,表現為升調節;大劑量或長期給藥,對性腺軸是降調節,對性腺起萎縮作用。
曲普瑞林多以短效注射劑為主,但是此類藥物半衰期很短,在體內極易降解,必須頻繁給藥才能取得療效。但是較長的治療周期會給醫生和患者帶來較大的不變,不能確保患者的依從性。因而,尋求適當的方法去延長曲普瑞林在體內的半衰期,減少給藥周期成為一種必然的趨勢。
高分子材料在藥物制劑領域的應用大大推動了藥物緩釋制劑的發展,尤其以生物可降解聚合物最為明顯。生物可降解聚合物是指一些能夠在水、酶作用下降解的高分子材料,常用的生物可降解聚合物包括淀粉、明膠、葡聚糖、白蛋白、聚乳酸、聚乳酸-羥基乙酸、聚鄰酯等。其中,PLGA因其與人體生物相容性良好且無毒無刺激的特點,在醫藥領域得到了廣泛的應用。目前,PLGA生物降解材料已被FDA批準用于藥物輸送系統,也使PLGA用于制備生物可降解型緩控釋給藥體系成為了國內外研究的熱點。
技術實現要素:
為此,本發明所要解決的技術問題在于現有技術中曲普瑞林制劑藥效時間短、載藥量低的問題,從而提出一種注射用曲普瑞林微球,并進一步公開其制備方法及用于制備曲普瑞林注射劑的用途。
為解決上述技術問題,本發明是通過以下技術方案來實現的:
本發明一種曲普瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75重量份,乙交脂丙交脂共聚物60-170重量份,羧甲基纖維素鈉20-40重量份,甘露醇70-100重量份,聚山梨脂801-4重量份。
本發明上述曲普瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75重量份,乙交脂丙交脂共聚物75重量份,羧甲基纖維素鈉30重量份,甘露醇85重量份,聚山梨脂802重量份。
本發明上述曲普瑞林微球,所述乙交脂丙交脂共聚物的分子量為25000~45000。
本發明上述曲普瑞林微球的制備方法,包括以下步驟:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為16.7~25.0%的溶液,備用;
稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為20.0~30.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解,制成質量濃度為7.0%~10.0%的甘露醇溶液、質量濃度為2.0%~4.0%的羧甲基纖維素鈉溶液、質量濃度為0.1~0.4%的聚山梨酯80溶液,并于80℃~90℃配制1.5~2.5小時,備用;
取聚乙烯醇加水,于80℃~90℃配制濃度為0.5%~1.5%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于12~18℃水浴冷卻;
(3)均質
將所述W1/O相產物以7.5~8.5mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
通過梯度升溫方法將均質后的藥液中含有的二氯甲烷去除;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并抽濾棄去溶液,即得所述微球。
本發明上述制備方法,包括以下步驟:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為16.7%的溶液,備用;
稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為25.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解,制成質量濃度為8.5%的甘露醇溶液、質量濃度為3.0%的羧甲基纖維素鈉溶液、質量濃度為0.2%的聚山梨酯80溶液,并于85℃配制2小時,備用;
取聚乙烯醇加水,于85℃配制濃度為1.0%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于15℃水浴冷卻;
(3)均質
將所述W1/O相產物以8mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
將均質后的藥液中含有的二氯甲烷去除;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并抽濾棄去溶液,即得所述微球。
本發明上述制備方法,所述步驟(5)中還包括對收集的所述微球進行冷凍干燥的步驟。
本發明上述制備方法,所述冷凍干燥的步驟具體為:
a、將擱板預凍至-30℃后,產品進箱;并繼續保持板溫-30℃至產品完全凍結;
b、控制板溫以4℃/小時的升溫速度升溫至-20℃,當達到-20℃以后開始抽真空至0.63mbar,繼續維持-20℃干燥10小時,然后以22.5℃/小時的升溫速度升溫至25℃后,抽真空至0.12mbar,并繼續維持25℃干燥6小時;
c、繼續抽真空至0.06mbar,控制溫度25℃保持繼續干燥4小時,結束。
本發明上述制備方法,所述步驟(4)中,所述去除二氯甲烷的步驟通過在真空條件下進行梯度升溫蒸發進行處理。
本發明上述制備方法,所述步驟(4)中,所述去除二氯甲烷的步驟具體為:
(1)升溫至10~15℃,抽真空至40~50kpa,保持10~20min;
(2)恒溫為10~15℃,繼續抽真空至30~40kpa,保持20~30min;
(3)升溫至25~30℃,繼續抽真空至30~40kpa,保持150~200min;
(4)升溫至35~45℃,繼續抽真空至30~40kpa,保持150~200min。
本發明上述微球或本發明上述方法制備得到的微球用于制備曲普瑞林注射劑的用途。
與現有技術相比,通過體外釋放度實驗、體內活性實驗、藥物代謝動力學實驗、加速穩定性實驗和長期穩定性實驗等的測定,表明本發明的一種曲普瑞林微球,延長了藥效時間、增大了載藥量,質量穩定、安全,處方工藝合理,符合藥用要求,臨床使用安全、可靠。
實驗例
以下實驗中以實施例制備的曲普瑞林微球作為供試品。其中,S1-1、S1-2、S1-3、S1-4、S1-5、S1-6、S1-7、S1-8、S1-9、S1-10和S1-11表示的分別為實施例1制備的不同批次的曲普瑞林微球,S2表示的為實施例2制備的曲普瑞林微球,S-1-1、S-1-2、S-1-3和S-1-4表示的分別為以實施例1的處方、實施例1的制備方法、不同分子量的PLGA制備的曲普瑞林微球,D表示的為現有技術中市售的曲普瑞林注射劑。
1、體外釋放度實驗
1.1測定方法
取實施例1、2制備的曲普瑞林微球1瓶,置50mL具塞試管中,加入釋放介質50mL,置于雙功能水浴恒溫振蕩器(50±1℃,60轉/分鐘)中,在1小時、7小時、24小時,72小時,120小時分別用注射器取出1mL,用0.45μm濾膜過濾,作為供試品溶液。精密吸取供試品溶液與對照品溶液各20μL,照高效液相色譜法(中國藥典2010年版二部附錄VD)測定。
1.2體外釋放度結果
對上述各供試品及對照品的體外釋放度的檢測數據見下表1。
表1體外釋放度實驗結果
1.3結論
由體外加速釋放的結果的可以得到,實施例1和實施例2制備的曲普瑞林微球體外釋放度結果相似,且體外緩釋期基本相同,均明顯優于現有技術的曲普瑞林注射劑的效果。
2、穩定性實驗
2.1實驗條件
2.1.1加速穩定性條件
溫度40±2℃,濕度75%±5%;溫度30℃±2℃,濕度65%±5%。
2.1.2長期穩定性條件
溫度:25℃±2℃,濕度60%±5%。
2.2實驗結果
對上述供試品和對照品的穩定性試驗的檢測數據見下表2所示。
表2穩定性實驗結果
2.3結論
由穩定性結果可知,實施例1制備的曲普瑞林微球在40℃和30℃加速條件下6個月與0個月相比基本穩定,25℃條件長期12個月與0個月相比基本穩定,釋放度結果相差不大,質量穩定,說明按本處方和工藝生產出的產品穩定良好,也說明處方的合理性及工藝的耐用性。
3、體內活性實驗
3.1實驗方法
取S1-1、S1-2、S1-3、S1-4、S1-5、S1-6和S1-7各一瓶,取S2兩瓶,分別用針頭刺入,用注射器分別加入懸浮用溶劑5mL,搖勻,選6只雄性成年SD大鼠,體重為320~400g,均分為兩組,每組3只,按給藥體積為0.05mL/100g,分別肌肉注射給藥1瓶。于注射后24小時采血,然后每周采血2次,連續1個月(直到29天或30天),用放射免疫法測老鼠血清中睪酮的濃度,不得大于1ng/mL。
3.2實驗結果
對上述供試品和對照品的體內活性檢測數據見下表3。
表3體內活性實驗結果(1)
體內活性實驗結果(1)的睪酮-時間曲線圖見附圖1。
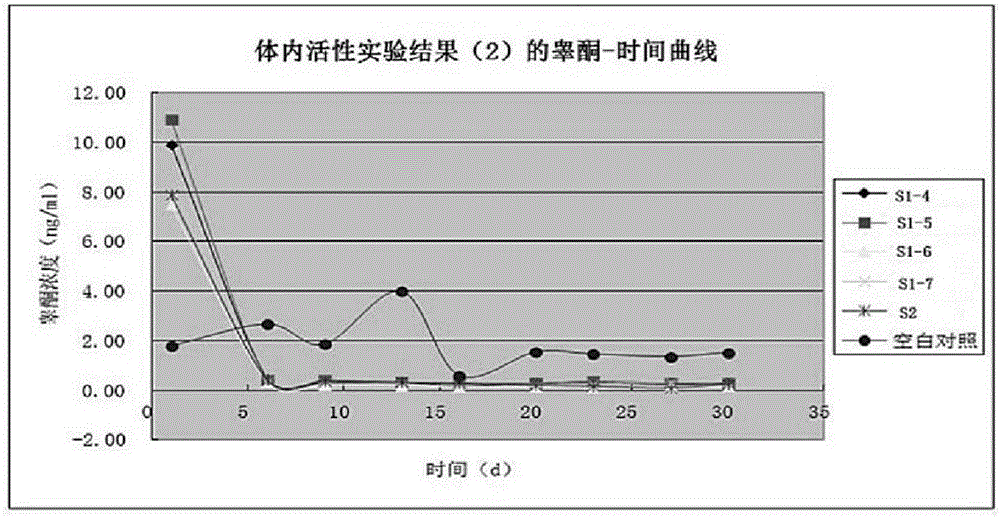
表3體內活性實驗結果(2)
體內活性實驗結果(2)的睪酮-時間曲線圖見附圖2。
3.3結論
由以上體內活性實驗的數據可知,實施例1和實施例2制備的曲普瑞林微球活性高度相似,均能將睪酮水平抑制在1ng/mg以下,且作用期均達到一個月。
4、藥物代謝動力學實驗
4.1實驗方法
委托上海藥物代謝研究中心進行試驗,實驗方法如下:12只比格犬隨機分成2組,每組6只,采用平行組試驗設計。比格犬肌肉注射3.75mg受試制劑1和受試制劑2后,收集給藥后0-35天不同時間點血漿樣本,采用液相色譜-串聯質譜(LC-MS/MS)法測定血漿中曲普瑞林的濃度,計算藥動學參數。
以實施例1制備的曲普瑞林微球作為受試制劑1,以實施例2制備的曲普瑞林微球作為受試制劑2。
4.2實驗結果
對上述制劑的藥物代謝動力學試驗的檢測結果見下表4。
表4藥物代謝動力學實驗結果(平均值±標準差,n=6)
比格犬肌肉注射曲普瑞林受試制劑1后,曲普瑞林在犬體內的血藥濃度達峰時間Tmax為0.25~1h(給藥后前2個采樣時間點)。達峰后血漿濃度快速下降,2-3天后血漿濃度降為0.3ng/mL。隨后藥物在體內再緩慢釋放,血漿濃度可維持在0.3~2ng/mL,直至給藥后30天,在給藥后第9~23天可觀察到二次達峰現象。
比格犬肌肉注射給予受試制劑2后,曲普瑞林在犬體內的血藥濃度達峰時間Tmax為0.25~1h。達峰后血漿濃度快速下降,于第6天后血漿濃度降為0.4ng/mL。隨后藥物在體內再緩慢釋放,血漿濃度可維持在0.1~2ng/mL,直至給藥后30天,在給藥后第16~19天可觀察到二次達峰現象。
4.3結論
受試制劑1給藥后,曲普瑞林在犬體內的達峰濃度Cmax為受試制劑2組的95.1%,相對生物利用度為102.8%。經統計檢驗,曲普瑞林的藥動學參數Cmax、AUC、MRT和t1/2在受試制劑1和受試制劑2間未表現出統計學差異(P>0.05)。
5、加速穩定性的動物體內活性實驗
5.1實驗方法
分別以S1-1、S1-2及S1-3在1、5、9、12、19、23、26、29及33小時取出的樣品進行加速穩定性實驗,在溫度40±2℃,濕度75%±5%;溫度30℃±2℃,濕度65%±5%的加速條件下放置,分別對各樣品在0、1、2、3、6個月取樣,進行大鼠藥效學的實驗,實驗方法如下:
取本品兩瓶,用注射器分別加入懸浮用溶劑5mL,搖勻,選6只雄性成年SD大鼠,體重為320~400g,均分為兩組,每組3只,按給藥體積為0.05mL/100g,分別肌肉注射給藥1瓶。于注射后24小時采血,然后每周采血2次,連續1個月(直到29天或30天),用放射免疫法測老鼠血清中睪酮的濃度,不得大于1ng/mL。
5.2實驗結果
對樣品的加速穩定性試驗測定數據見下表5-10。
表5 S1-1的40℃加速穩定性實驗結果(單位ng/mL)
S1-1的30℃加速穩定性實驗的睪酮-時間曲線圖見附圖3。
表6 S1-1的30℃加速穩定性實驗結果(單位ng/mL)
S1-2的40℃加速穩定性實驗的睪酮-時間曲線圖見附圖4。
表7 S1-2的40℃加速穩定性實驗結果(單位ng/mL)
S1-2的30℃加速穩定性實驗的睪酮-時間曲線圖見附圖5。
表8 S1-2的30℃加速穩定性實驗結果(單位ng/mL)
S1-2的30℃加速穩定性實驗的睪酮-時間曲線圖見附圖6。
表9 S1-3的40℃加速穩定性實驗結果(單位ng/mL)
S1-3的30℃加速穩定性實驗的睪酮-時間曲線圖見附圖7。
表10 S1-3的30℃加速穩定性實驗結果(單位ng/mL)
S1-3的30℃加速穩定性實驗的睪酮-時間曲線圖見附圖8。
5.3結論
通過加速穩定性的動物體內活性的實驗結果可知,實施例1制備的曲普瑞林微球的性質穩定,處方合理,工藝耐用。
6、長期穩定性的動物體內活性實驗
6.1實驗方法
將樣品在溫度:25℃±2℃,濕度60%±10%;條件下放置,分別在0、3、6、9、12、18個月取樣,進行大鼠藥效學的實驗,實驗方法如下:
取本品兩瓶,用注射器分別加入懸浮用溶劑5mL,搖勻,選6只雄性成年SD大鼠,體重為320~400g,均分為兩組,每組3只,按給藥體積為0.05mL/100g,分別肌肉注射給藥1瓶。于注射后24小時采血,然后每周采血2次,連續1個月(直到29天或30天),用放射免疫法測老鼠血清中睪酮的濃度,不得大于1ng/mL。
6.2實驗結果
對上述樣品的長期穩定性實驗結果見表11-13。
表11 S1-1的25℃長期穩定性實驗結果(單位ng/mL)
S1-1的25℃長期穩定性實驗的睪酮-時間曲線圖見附圖9。
表12 S1-2的25℃長期穩定性實驗結果(單位ng/mL)
S1-2的25℃長期穩定性實驗的睪酮-時間曲線圖見附圖10。
表13 S13的25℃長期穩定性實驗結果(單位ng/mL)
S1-3的25℃長期穩定性實驗的睪酮-時間曲線圖見附圖11。
6.3結論
通過長期穩定性的動物體內活性的實驗結果可知,實施例1制備的曲普瑞林微球的性質穩定,處方合理,工藝耐用。
7、PLGA分子量的篩選實驗
PLGA作為構成球體的骨架材料,其分子量的大小,對微球釋放行為的影響較大。
以體外釋放度作為考察指標,篩選了四種不同分子量(11000,25000,33000,58000)的PLGA,并與原研制品釋放度進行相似性擬合,不同PLGA分子量的篩選實驗結果見表14、圖12、圖13、圖14和圖15。
表14不同PLGA分子量的體外釋放度結果
結論:(1)分子量為11000的PLGA所制備的微球釋放過快;(2)分子量為58000的PLGA所制備的微球前期和中期釋放過慢;(3)分子量為33000的PLGA所制備的微球釋放度前期和中期釋放較慢;(4)分子量為25000的PLGA所制備的微球釋放度與S2相似度較高,體外釋放度相當。
8、PLGA用量的篩選實驗
以體外釋放度作為主要考察指標,篩選不同PLGA用量的處方,探究PLGA用量對處方組成的影響,所篩選的處方見表15,不同處方的體外釋放度結果結果見表15和圖16。
表15所篩選的不同PLGA用量的處方
表16不同PLGA用量的處方的體外釋放度結果
結果分析:(1)原料損失:原料藥為水溶性藥物制,制備過程中易溶于外水相產生損失,故在生產中要存在過量加入現象,需根據球體載藥量計算原料損失百分比;(2)由于原料藥制備過程中損失,所以需根據球體載藥量確定單位劑量實際構成球體的輔料用量;(3)體外釋放度:處方2與S2相似度較高,其余三組前期釋放均較慢。
結論:處方2體外釋放度與S2較相似,故選用處方2作為PLGA的投料量,并據此進行重復試驗,確定載藥量的穩定性,以載藥量確定單位劑量產品的PLGA處方量和原料藥投料量。
附圖說明
為了使本發明的內容更容易被清楚地理解,下面根據本發明的具體實施例并結合附圖,對本發明作進一步詳細地說明,其中:
圖1是實驗例中體內活性實驗結果(1)的睪酮-時間曲線圖;
圖2是實驗例中體內活性實驗結果(2)的睪酮-時間曲線圖;
圖3是實驗例5中S1-1的40℃加速穩定性實驗的睪酮-時間曲線圖;
圖4是實驗例5中S1-1的30℃加速穩定性實驗的睪酮-時間曲線圖;
圖5是實驗例5中S1-2的40℃加速穩定性實驗的睪酮-時間曲線圖;
圖6是實驗例5中S1-2的30℃加速穩定性實驗的睪酮-時間曲線圖;
圖7是實驗例5中S1-3的40℃加速穩定性實驗的睪酮-時間曲線圖;
圖8是實驗例5中S1-3的30℃加速穩定性實驗的睪酮-時間曲線圖;
圖9是實驗例6中S1-1的25℃長期穩定性實驗的睪酮-時間曲線圖;
圖10是實驗例6中S1-2的25℃長期穩定性實驗的睪酮-時間曲線圖;
圖11是實驗例6中S1-3的25℃長期穩定性實驗的睪酮-時間曲線圖;
圖12是實驗例7中S-1-1的體外釋放度結果曲線圖;
圖13是實驗例7中S-1-2的體外釋放度結果曲線圖;
圖14是實驗例7中S-1-3和S-1-4的體外釋放度結果曲線圖;
圖15是實驗例7中S1-8、S1-9、S1-10和S1-11的體外釋放度結果曲線圖;
圖16是實驗例8中不同PLGA用量的處方的體外釋放度結果曲線圖。
具體實施方式
涉及試劑
聚乳酸羥基乙酸共聚物(PLGA):Lakeshore,美國;
醋酸曲普瑞林(Triptorelin):杭州中肽生化有限公司;
二氯甲烷(DCM):AR,北京化工;
甲醇:AR北京化工;羧甲基纖維素鈉(CMC-Na):安徽山河藥用輔料;
甘露醇:廣西南寧化學制藥有限公司;聚乙烯醇:斯百全試劑;
聚山梨酯80南京威爾化工有限公司;純化水。
涉及儀器
電子天平:sartorius BP211D,德國;振蕩乳化機:minibeadbeater,美國;
進樣泵:Harvord Plusll,美國;均質機:silveron均質機,英國;
旋轉蒸發儀:RE5202,上海亞榮;凍干機:Chirist 2-10D,德國。
實施例1
本實施例所述曲譜瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75g,乙交脂丙交脂共聚物(分子量為25000)75g,羧甲基纖維素鈉30g,甘露醇85g,聚山梨脂802g。
本實施例一種曲譜瑞林微球由以下方法制備:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為20.0%的溶液,備用;稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為25.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解制成質量濃度為8.5%的甘露醇溶液、質量濃度為3.0%的羧甲基纖維素鈉溶液、質量濃度為0.2%的聚山梨酯80溶液,并于85℃配制2小時,備用;
取聚乙烯醇加水,于85℃配制濃度為1.0%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于15℃水浴冷卻;
(3)均質
將所述W1/O相產物以8.0mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
通過梯度升溫、攪拌和抽真空對均質后的藥液內的二氯甲烷進行揮發;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并用1μm濾膜抽濾裝置進行抽濾棄去溶液,即得所述微球;
(6)冷凍干燥
a、將擱板預凍至-30℃后,產品進箱;并繼續保持板溫-30℃至產品完全凍結;
b、控制板溫以4℃/小時的升溫速度升溫至-20℃,當達到-20℃以后開始抽真空至0.63mbar,繼續維持-20℃干燥10小時,然后以22.5℃/小時的升溫速度升溫至25℃后,再次開始抽真空至0.12mbar,并繼續維持25℃干燥6小時;
c、繼續抽真空至0.06mbar,控制溫度25℃保持繼續干燥4小時,結束。
實施例2
本實施例一種曲普瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75g,乙交脂丙交脂共聚物(分子量為45000)170g,羧甲基纖維素鈉30g,甘露醇85g,聚山梨脂802g。
本實施例一種曲譜瑞林微球由以下方法制備:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為16.7%的溶液,備用;
稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為25.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解制成質量濃度為8.5%的甘露醇溶液、質量濃度為3.0%的羧甲基纖維素鈉溶液、質量濃度為0.2%的聚山梨酯80溶液,并于85℃配制2小時,備用;
取聚乙烯醇加水,于85℃配制濃度為1.0%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于15℃水浴冷卻;
(3)均質
將所述W1/O相產物以8.0mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
通過梯度升溫、攪拌和抽真空對均質后的藥液內的二氯甲烷進行揮發;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并用1μm濾膜抽濾裝置進行抽濾棄去溶液,即得所述微球;
(6)冷凍干燥
a、將擱板預凍至-30℃后,產品進箱;并繼續保持板溫-30℃至產品完全凍結;
b、控制板溫以4℃/小時的升溫速度升溫至-20℃,當達到-20℃以后開始抽真空至0.63mbar,繼續維持-20℃干燥10小時,然后以22.5℃/小時的升溫速度升溫至25℃后,再次開始抽真空至0.12mbar,并繼續維持25℃干燥6小時;
c、繼續抽真空至0.06mbar,控制溫度25℃保持繼續干燥4小時,結束。
實施例3
本實施例一種曲譜瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75g,乙交脂丙交脂共聚物(分子量為25000)60g,羧甲基纖維素鈉40g,甘露醇70g,聚山梨脂804g。
本實施例一種曲譜瑞林微球由以下方法制備:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為20.0%的溶液,備用;
稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為20.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解制成質量濃度為7.0%的甘露醇溶液、質量濃度為4.0%的羧甲基纖維素鈉溶液、質量濃度為0.4%的聚山梨酯80溶液,并于80℃配制2.5小時,備用;
取聚乙烯醇加水,于80℃配制濃度為1.5%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于12℃水浴冷卻;
(3)均質
將所述W1/O相產物以8.5mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
通過梯度升溫、攪拌和抽真空對均質后的藥液內的二氯甲烷進行揮發;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并用1μm濾膜抽濾裝置進行抽濾棄去溶液,即得所述微球;
(6)冷凍干燥
a、將擱板預凍至-30℃后,產品進箱;并繼續保持板溫-30℃至產品完全凍結;
b、控制板溫以4℃/小時的升溫速度升溫至-20℃,當達到-20℃以后開始抽真空至0.63mbar,繼續維持-20℃干燥10小時,然后以22.5℃/小時的升溫速度升溫至25℃后,再次開始抽真空至0.12mbar,并繼續維持25℃干燥6小時;
c、繼續抽真空至0.06mbar,控制溫度25℃保持繼續干燥4小時,結束。
實施例4
本實施例一種曲譜瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75g,乙交脂丙交脂共聚物(分子量為25000)170g,羧甲基纖維素鈉20g,甘露醇100g,聚山梨脂801g。
本實施例一種曲譜瑞林微球由以下方法制備:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為25.0%的溶液,備用;
稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為30.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解制成質量濃度為10.0%的甘露醇溶液、質量濃度為2.0%的羧甲基纖維素鈉溶液、質量濃度為0.1%的聚山梨酯80溶液,并于90℃配制1.5小時,備用;
取聚乙烯醇加水,于90℃配制濃度為0.5%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于18℃水浴冷卻;
(3)均質
將所述W1/O相產物以7.5mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
通過梯度升溫、攪拌和抽真空對均質后的藥液內的二氯甲烷進行揮發;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并用1μm濾膜抽濾裝置進行抽濾棄去溶液,即得所述微球;
(6)冷凍干燥
a、將擱板預凍至-30℃后,產品進箱;并繼續保持板溫-30℃至產品完全凍結;
b、控制板溫以4℃/小時的升溫速度升溫至-20℃,當達到-20℃以后開始抽真空至0.63mbar,繼續維持-20℃干燥10小時,然后以22.5℃/小時的升溫速度升溫至25℃后,再次開始抽真空至0.12mbar,并繼續維持25℃干燥6小時;
c、繼續抽真空至0.06mbar,控制溫度25℃保持繼續干燥4小時,結束。
實施例5
本實施例一種曲譜瑞林微球,由以下重量份數的組分組成:醋酸曲普瑞林3.75g,乙交脂丙交脂共聚物(分子量為25000)75g,羧甲基纖維素鈉30g,甘露醇80g,聚山梨脂802g。
本實施例一種曲譜瑞林微球由以下方法制備:
(1)溶液配制
稱取處方量的醋酸曲普瑞林溶解于水中,制成質量濃度為25.0%的溶液,備用;
稱取處方量的乙交脂丙交脂共聚物,溶于二氯甲烷配制成質量濃度為25.0%的溶液,備用;
取處方量的甘露醇、羧甲基纖維素鈉、聚山梨酯80混勻,加水溶解制成質量濃度為8.5%的甘露醇溶液、質量濃度為3.0%的羧甲基纖維素鈉溶液、質量濃度為0.2%的聚山梨酯80溶液,并于85℃配制2小時,備用;
取聚乙烯醇加水,于85℃配制濃度為1.0%的水溶液,備用;
(2)乳化
將所述乙交脂丙交脂共聚物溶液加入至所述醋酸曲普瑞林溶液中,振蕩乳化,制備成W1/O相產物,并于15℃水浴冷卻;
(3)均質
將所述W1/O相產物以8.0mL/min注射速度注射入所述聚乙烯醇溶液中均質處理;
(4)溶劑蒸發
通過梯度升溫、攪拌和抽真空對均質后的藥液內的二氯甲烷進行揮發;
(5)微球收集
將去除二氯甲烷的藥液進行冷凍離心,并用1μm濾膜抽濾裝置進行抽濾棄去溶液,即得所述微球;
(6)冷凍干燥
a、將擱板預凍至-30℃后,產品進箱;并繼續保持板溫-30℃至產品完全凍結;
b、控制板溫以4℃/小時的升溫速度升溫至-20℃,當達到-20℃以后開始抽真空至0.63mbar,繼續維持-20℃干燥10小時,然后以22.5℃/小時的升溫速度升溫至25℃后,再次開始抽真空至0.12mbar,并繼續維持25℃干燥6小時;
c、繼續抽真空至0.06mbar,控制溫度25℃保持繼續干燥4小時,結束。
顯然,上述實施例僅僅是為清楚地說明所作的舉例,而并非對實施方式的限定。對于所屬領域的普通技術人員來說,在上述說明的基礎上還可以做出其它不同形式的變化或變動。這里無需也無法對所有的實施方式予以窮舉。而由此所引伸出的顯而易見的變化或變動仍處于本發明創造的保護范圍之中。
- 還沒有人留言評論。精彩留言會獲得點贊!