一種藥物緩釋水凝膠及其制備方法與流程
本發明涉及高分子技術領域及藥物緩釋型技術領域,具體涉及的是一種藥物緩釋水凝膠及其制備方法。
背景技術:
水凝膠是一類以水溶性高分子聚合物為基質骨架材料的交聯聚合物,具有載藥量大,保濕性強,與皮膚的相容性好,耐老化等特點,因此水凝膠可廣泛應用在組織修復、人造器官、藥物緩釋及傳感器等諸多領域。常用作藥物緩釋載體的水凝膠貼可以反復揭貼、隨時終止給藥;且保證劑量準確,血藥濃度平衡無峰谷現象,可減少毒副作用;在工業生產中無有機溶媒污染,符合環保要求,從而成為當代藥劑學研究的熱門之一。
水凝膠的藥物釋放過程是一個動力學和熱力學的復合過程,為了讓水凝膠的釋放作用更加穩定,通常加入粉料來延長產品的擴散通道,穩定藥物釋放的過程,例如日本藥店販售的水凝膠藥劑就有規定加入高嶺土用于穩定藥物釋放的過程。但是,現有的水凝膠仍存在緩釋效果差、不穩定等問題,使得有效成分不能發揮應有的藥效,因此,水凝膠的藥物釋放的穩定性問題仍有進一步探討的空間。
技術實現要素:
本發明的目的在于提供一種藥物緩釋水凝膠及其制備方法,制備的水凝膠緩釋效果好,藥物釋放的穩定性有所提高。
為了達成上述目的,本發明的解決方案是:
一種藥物緩釋水凝膠,包括如下重量百分比的原料:
所述促滲劑為月桂氮酮或者二甲亞砜,所述交聯劑為甘羥鋁,所述增粘劑為聚乙烯吡咯烷酮,所述催化劑為酒石酸。
所述藥物分子為吲哚美辛或者布洛芬。
所述丙烯酸鈉樹脂由日本昭和公司購買得到。
所述溶劑為乙醇和丙三醇的混合物,所述乙醇的重量百分用量為1~5%,所述丙三醇的重量百分用量為5~40%。
該藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)將0.2~2%的馬來酸酐十四醇基丙磺酸鈉、6~45%的溶劑、1~5%的促滲劑以及0.1~1%的藥物分子混合均勻,形成單體包裹藥物溶液;
(2)將0.01~0.5%的催化劑酒石酸溶于水中制備成酒石酸水溶液;
(3)向步驟(1)得到的所述單體包裹藥物溶液中依次加入1~15%的丙烯酸鈉樹脂、0.01~1%的交聯劑甘羥鋁、1~15%的增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將所述水凝膠前驅物模壓成型,在60Coγ射線下輻照后取出,所述60Coγ射線的輻照量為0.5-8kGy,即得該藥物緩釋水凝膠產品;步驟(1)~(3)中各組分的用量總和為100%。
步驟(1)中,所述溶劑為乙醇和丙三醇的混合物,所述乙醇的重量百分用量為1~5%,所述丙三醇的重量百分用量為5~40%。
馬來酸酐十四醇基丙磺酸鈉是經典的無皂乳液聚合中的反應性高分子乳化單體【Acta Polymerica,1998,29(12):4508-4515】,具有良好的乳化能力,在本發明的技術方案中,將其作為自乳化單體,先與藥物分子、溶劑和促滲劑共混,生成有表面活性的單體包裹藥物后,將該單體包裹藥物在由丙烯酸鈉樹脂、交聯劑甘羥鋁、增粘劑聚乙烯吡咯烷酮以及酒石酸水溶液構成的水凝膠基質中進行輻照聚合,通過輻照使得馬來酸酐十四醇基丙磺酸鈉交聯形成膠束粒子,從而獲得穩定的膠束粒子包裹的緩釋藥物水凝膠,由此提高藥物釋放的穩定性。
附圖說明
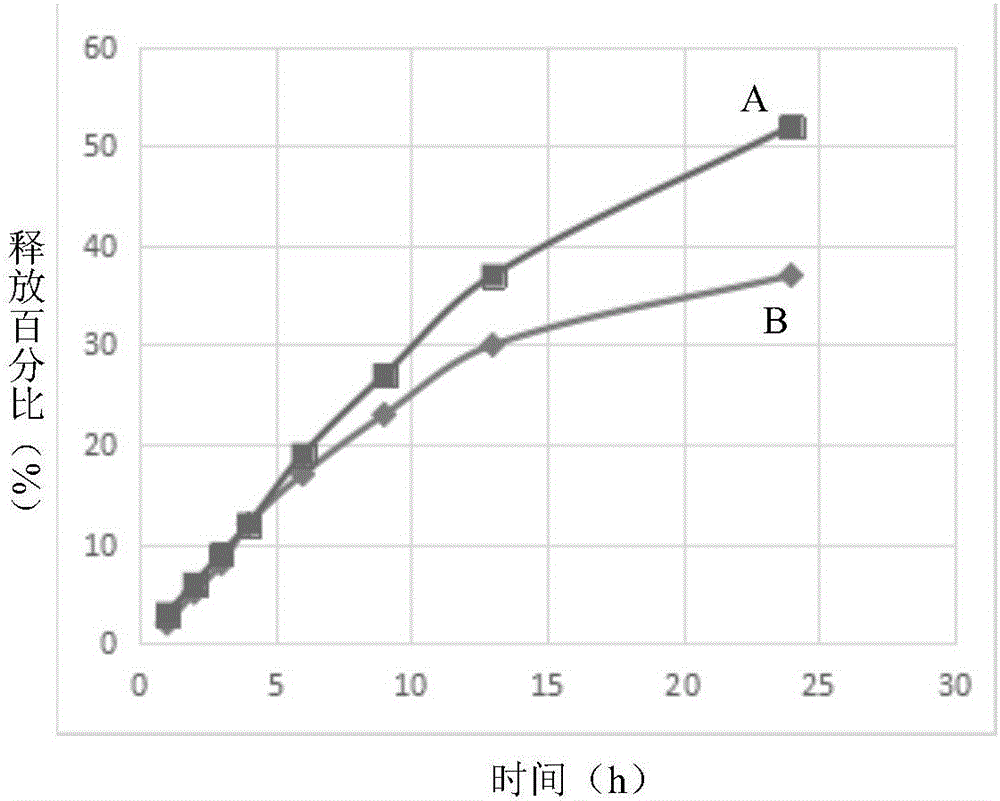
圖1為本發明實施例一中制備得到的藥物緩釋水凝膠(A)與市售水凝膠巴布貼(B)的緩釋效果對比圖。
具體實施方式
為了進一步解釋本發明的技術方案,下面通過具體實施例來對本發明進行詳細闡述。
一種藥物緩釋水凝膠,包括如下重量百分比的原料:
所述促滲劑為月桂氮酮或者二甲亞砜,所述交聯劑為甘羥鋁,所述增粘劑為聚乙烯吡咯烷酮,所述催化劑為酒石酸。
所述藥物分子為吲哚美辛或者布洛芬。
所述丙烯酸鈉樹脂由日本昭和公司購買得到。
所述溶劑為乙醇和丙三醇的混合物,所述乙醇的重量百分用量為1~5%,所述丙三醇的重量百分用量為5~40%。
一種藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)將0.2~2%的馬來酸酐十四醇基丙磺酸鈉、6~45%的溶劑、1~5%的促滲劑以及0.1~1%的藥物分子混合均勻,形成單體包裹藥物溶液;
(2)將0.01~0.5%的催化劑酒石酸溶于水中制備成酒石酸水溶液;
(3)向步驟(1)得到的所述單體包裹藥物溶液中依次加入1~15%的丙烯酸鈉樹脂、0.01~1%的交聯劑甘羥鋁、1~15%的增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將所述水凝膠前驅物模壓成型,在60Coγ射線下輻照后取出,所述60Coγ射線的輻照量為0.5-8kGy,即得該藥物緩釋水凝膠產品;步驟(1)~(3)中各組分的用量總和為100%。
實施例一
該藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)按照配方比例,將0.5g馬來酸酐十四醇基丙磺酸鈉、3g乙醇、15g丙三醇、0.35g吲哚美辛以及2g促滲劑月桂氮酮混合均勻,形成單體包裹藥物溶液;
(2)將0.1g催化劑酒石酸溶于64.03g水中制備成酒石酸水溶液;
(3)向步驟(1)得到的單體包裹藥物溶液中依次加入10g丙烯酸鈉樹脂、0.02g交聯劑甘羥鋁、5g增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將水凝膠前驅物模壓成片狀,在60Coγ射線下輻照后取出,60Coγ射線的輻照量為4kGy,即得該藥物緩釋水凝膠產品。
為了更好地證明該藥物緩釋水凝膠的緩釋效果,將該藥物緩釋水凝膠(A)與市售水凝膠巴布貼(B)進行了對比,兩者的藥物分子的含量相同,結果如圖1所示,其中x軸為時間(h),y軸為釋放百分比(%)。結果表明:該藥物緩釋水凝膠產品的緩釋效果和穩定性都較佳。
實施例二
該藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)按照配方比例,將0.8g馬來酸酐十四醇基丙磺酸鈉、3g乙醇、20g丙三醇、0.5g吲哚美辛以及2.5g促滲劑月桂氮酮混合均勻,形成單體包裹藥物溶液;
(2)將0.1g催化劑酒石酸溶于55.08g水中制備成酒石酸水溶液;
(3)向步驟(1)得到的單體包裹藥物溶液中依次加入10g丙烯酸鈉樹脂、0.02g交聯劑甘羥鋁、8g增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將水凝膠前驅物模壓成片狀,在60Coγ射線下輻照后取出,60Coγ射線的輻照量為0.5kGy,即得該藥物緩釋水凝膠產品。
實施例三
該藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)按照配方比例,將2g馬來酸酐十四醇基丙磺酸鈉、3g乙醇、10g丙三醇、0.5g吲哚美辛以及3g促滲劑二甲亞砜混合均勻,形成單體包裹藥物溶液;
(2)將0.1g催化劑酒石酸溶于54.35g水中制備成酒石酸水溶液;
(3)向步驟(1)得到的單體包裹藥物溶液中依次加入15g丙烯酸鈉樹脂、0.05g交聯劑甘羥鋁、12g增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將水凝膠前驅物模壓成片狀,在60Coγ射線下輻照后取出,60Coγ射線的輻照量為2kGy,即得該藥物緩釋水凝膠產品。
實施例四
該藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)按照配方比例,將1.5g馬來酸酐十四醇基丙磺酸鈉、5g乙醇、20g丙三醇、0.5g布洛芬以及2g促滲劑月桂氮酮混合均勻,形成單體包裹藥物溶液;
(2)將0.1g催化劑酒石酸溶于50.7g水中制備成酒石酸水溶液;
(3)向步驟(1)得到的單體包裹藥物溶液中依次加入15g丙烯酸鈉樹脂、0.2g交聯劑甘羥鋁、5g增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將水凝膠前驅物模壓成片狀,在60Coγ射線下輻照后取出,60Coγ射線的輻照量為6kGy,即得該藥物緩釋水凝膠產品。
實施例五
該藥物緩釋水凝膠的制備方法,包括以下步驟:
(1)按照配方比例,將1g馬來酸酐十四醇基丙磺酸鈉、3g乙醇、15g丙三醇、0.5g吲哚美辛以及4g促滲劑二甲亞砜混合均勻,形成單體包裹藥物溶液;
(2)將0.4g催化劑酒石酸溶于54.3g水中制備成酒石酸水溶液;
(3)向步驟(1)得到的單體包裹藥物溶液中依次加入12g丙烯酸鈉樹脂、0.8g交聯劑甘羥鋁、9g增粘劑聚乙烯吡咯烷酮以及步驟(2)得到的酒石酸水溶液,混合均勻,得到稠狀的水凝膠前驅物;
(4)將水凝膠前驅物模壓成片狀,在60Coγ射線下輻照后取出,60Coγ射線的輻照量為8kGy,即得該藥物緩釋水凝膠產品。
上述實施例和圖式并非限定本發明的產品形態和式樣,任何所屬技術領域的普通技術人員對其所做的適當變化或修飾,皆應視為不脫離本發明的專利范疇。
- 還沒有人留言評論。精彩留言會獲得點贊!