具有改善的儲存穩定性的藥物組合物及其制備方法與流程
本發明涉及具有改善的儲存穩定性的藥物組合物及其制備方法,更具體地涉及包含兩親性嵌段共聚物的水溶性差的藥物的藥物組合物,其中特定相關化合物的含量保持在規定限量以內;并且涉及其制備方法。
背景技術:
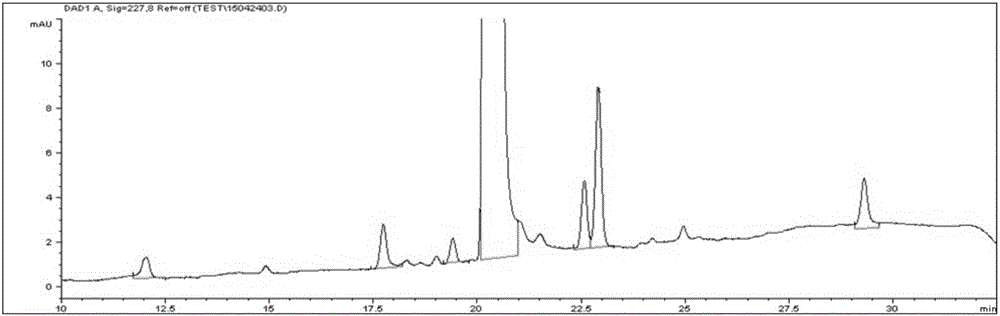
:水溶性差的藥物的溶解是經由口服或腸胃外給藥將藥物遞送到體內的關鍵技術。這種溶解方法包括在水溶液中添加表面活性劑以形成膠束,然后在其中包埋(entrapping)水溶性差的藥物的方法。用作表面活性劑的兩親性嵌段共聚物包含親水性聚合物嵌段和疏水性聚合物嵌段。由于親水性聚合物嵌段在體內直接接觸血液蛋白質和細胞膜,因此已使用具有生物相容性的聚乙二醇或單甲氧基聚乙二醇等。疏水性聚合物嵌段改善與疏水性藥物的親和性,因此已使用具有可生物降解的聚丙交酯、聚乙交酯、聚(乳酸-乙交酯)(poly(lactic-glycolide))、聚己內酯、聚氨基酸或聚原酸酯等。特別地,因為聚丙交酯衍生物具有優異的生物相容性并且在體內水解為無害的乳酸,因此它們已經以各種形式應用于藥物載體。聚丙交酯衍生物隨其分子量變化具有不同的物理性質,并且已經開發出各種形式,例如微球、納米顆粒、聚合物凝膠和植入劑。第6,322,805號美國專利公開了用于遞送水溶性差的藥物的組合物,其由聚合物膠束型藥物載體和水溶性差的藥物組成,其中所述聚合物膠束型藥物載體由二嵌段共聚物或三嵌段共聚物形成,所述二嵌段共聚物或三嵌段共聚物不由交聯劑交聯,其由選自聚丙交酯、聚乙交酯、聚(丙交酯-乙交酯)、聚己內酯及其衍生物的至少一種可生物降解的疏水性聚合物和聚(環氧烷)作為親水性聚合物組成,其中將水溶性差的藥物物理地包埋在藥物載體中并溶解,并且其中所述聚合物膠束型藥物載體在水中形成透明水溶液并將水溶性差的藥物有效地遞送到體內。根據上述美國專利,聚乙二醇-聚丙交酯二嵌段共聚物是通過以下步驟合成的:從單甲氧基聚乙二醇中除去水分,向其中加入溶解在甲苯中的辛酸亞錫并減壓除去甲苯,向所得混合物中加入D,L-丙交酯并進行聚合反應,加入氯仿以溶解所產生的嵌段共聚物,在攪拌下小份地滴加過量的乙醚以形成沉淀,過濾形成的沉淀,并用乙醚洗滌數次。然而,該方法難以在大規模生產中使用,因此不可商用。此外,用于純化的醚可能殘留在最終的聚合物膠束組合物中。第8,853,351號美國專利公開了一種制備兩親性嵌段共聚物的方法,其包括(a)將兩親性嵌段共聚物溶解在水混溶性有機溶劑中;(b)向步驟(a)中獲得的聚合物溶液中加入并混合堿金屬鹽(碳酸氫鈉、碳酸鈉、碳酸氫鉀、碳酸鉀或碳酸鋰)的水溶液;(c)通過鹽析對步驟(b)中獲得的溶液進行有機相和水相分離;和(d)分離步驟(c)中獲得的有機相并除去其中的有機溶劑以回收聚合物。然而,該方法涉及復雜的步驟,需要額外的步驟以除去堿金屬鹽和用于鹽析的鹽(氯化鈉或氯化鉀),并且即使在將其除去后也可能具有殘留的金屬鹽。必須在各個方面嚴格控制藥物的雜質。特別地,在雜質源于活性藥物成分(API)的情況下,每個國家在其藥物審批指南中確定了藥物產品中源自API的、已知或未知雜質(相關化合物)的量的上限。此外,還有一些國際上使用的標準,ICH指南Q3A是有代表性的一個。在該指南中,在審批藥物時,藥物中每種相關化合物的量限制為最多0.1%或0.2%等,并且根據超過限量的相關化合物,有差別地適用應當規定的諸如毒性相關數據等的信息。這意味著在制備藥物的過程中必須減少相關化合物的量,因為藥物的相關化合物在體內如何作用是未知的。因此,用于減少相關化合物的制造方法和根據每種相關化合物的特性(結構和毒性)設定其量的上限是藥物質量控制中的必要因素。技術實現要素:技術問題本發明的一個目的為提供含有兩親性嵌段共聚物的水溶性差的藥物的聚合物膠束型藥物組合物,其含有在規定限量以內的量的特定相關化合物。本發明的另一個目的為提供制備所述藥物組合物的方法。技術手段本發明的一方面提供了聚合物膠束藥物組合物,其包含:包含親水性嵌段(A)和疏水性嵌段(B)的純化的兩親性嵌段共聚物,以及一種或多種水溶性差的藥物,其選自紫杉醇和多西紫杉醇,其中當所述藥物組合物在40℃下儲存6個月時,其含有以所述水溶性差的藥物的初始量為100重量份計的0.2重量份以下的由下式1表示的相關化合物:[式1]其中:R1為H或COCH3,R2為苯基或O(CH3)3。本發明的另一方面提供了制備聚合物膠束藥物組合物的方法,其包括:(a)純化包含親水性嵌段(A)和疏水性嵌段(B)的兩親性嵌段共聚物;(b)將選自紫杉醇和多西紫杉醇的一種或多種水溶性差的藥物和純化的兩親性嵌段共聚物溶解在有機溶劑中;和(c)向步驟(b)中獲得的溶液中加入水性溶劑以形成聚合物膠束;當所述藥物組合物在40℃下儲存6個月時,其含有以所述水溶性差的藥物的初始量為100重量份計的0.2重量份以下的由上式1表示的相關化合物。有益效果根據本發明,可以獲得具有減少的相關化合物和改善的儲存穩定性的水溶性差的藥物的藥物組合物。附圖說明圖1為對實驗例1中使用的、經過六個月加速試驗的、含有紫杉醇的聚合物膠束組合物進行HPLC分析所得的色譜圖。圖2顯示對實驗例1中獲得的相關化合物(RRT0.96±0.02(0.94~0.98),其在下文中可與RRT0.96互換使用)進行LC/MS/MS分析的產物離子掃描結果。圖3顯示對實驗例2中通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質進行LC/MS/MS分析的結果。圖4顯示對實驗例2中通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質進行LC/MS/MS分析的產物離子掃描結果,以及聚合物膠束藥物組合物的結果:(a)經過六個月加速試驗的聚合物膠束藥物組合物樣品的分析結果;(b)通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質的分析結果。圖5顯示對實驗例2中通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質進行NMR分析的1HNMR分析結果。圖6顯示對實驗例2中通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質進行NMR分析的13CNMR分析結果。圖7顯示對實驗例2中通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質進行NMR分析的COSY(相關光譜)分析結果。圖8顯示對本發明實驗例2中通過熱分解紫杉醇所得的混合物中在RRT0.96處所得物質進行NMR分析的HMBC(異核多鍵相關光譜)分析結果。圖9是在實驗例5中進行HPLC分析所得的色譜圖。具體實施方式下面對本發明進行更詳細地說明。本發明實施方案的藥物組合物包含含有親水性嵌段(A)和疏水性嵌段(B)的純化的兩親性嵌段共聚物。根據本發明的一個實施方案,所述兩親性嵌段共聚物包含由親水性嵌段(A)和疏水性嵌段(B)組成的A-B型二嵌段共聚物或B-A-B型三嵌段共聚物。根據本發明的一個實施方案,所述兩親性嵌段共聚物可包含,以該共聚物的總重量計,20-95重量%、更具體地40-95重量%的親水性嵌段。此外,所述兩親性嵌段共聚物可包含,以該共聚物的總重量計,5-80重量%、更具體地5-60重量%的疏水性嵌段。根據本發明的一個實施方案,所述兩親性嵌段共聚物的數均分子量可以為1,000-50,000道爾頓,更具體地為1,500-20,000道爾頓。根據本發明的一個實施方案,所述親水性嵌段是具有生物相容性的聚合物并且可包含選自聚乙二醇或其衍生物、聚乙烯吡咯烷酮、聚乙烯醇、聚丙烯酰胺及其組合中的一種或多種,更具體地,其可包含選自聚乙二醇、單甲氧基聚乙二醇及其組合中的一種或多種。親水性嵌段的數均分子量可以為200-20,000道爾頓,更具體地為200-10,000道爾頓。根據本發明的一個實施方案,所述疏水性嵌段是具有生物可降解性的聚合物,并且可以為衍生自α-羥基酸的單體的聚合物。具體地,其可包含選自聚丙交酯、聚乙交酯、聚扁桃酸、聚己內酯、聚二噁烷-2-酮、聚氨基酸、聚原酸酯、聚酐、聚碳酸酯及其組合的一種或多種,更具體地,其可包含選自聚丙交酯、聚乙交酯、聚己內酯、聚二噁烷-2-酮及其組合中的一種或多種。疏水性嵌段的數均分子量可以為200-20,000道爾頓,更具體地為200-10,000道爾頓。根據本發明的一個實施方案,包含聚(α-羥基酸)疏水性聚合物嵌段的兩親性嵌段共聚物可以通過已知的開環聚合方法合成,所述方法使用具有羥基的親水性聚合物作為引發劑和α-羥基酸內酯單體。例如,以具有羥基的親水性聚乙二醇或單甲氧基聚乙二醇為引發劑,L-丙交酯或D,L-丙交酯可以通過開環聚合。根據作為引發劑的親水性嵌段中存在的羥基的數目,可以合成二嵌段共聚物或三嵌段共聚物。在開環聚合中,可以使用有機金屬催化劑,例如氧化錫、氧化鉛、辛酸錫、辛酸銻等,在制備醫療用聚合物時優選使用具有生物相容性的辛酸錫。在本發明的實施方案中,使用純化的共聚物作為兩親性嵌段共聚物。根據本發明的優選實施方案,兩親性嵌段共聚物是已通過升華純化的共聚物。升華提純可以在優選80-120℃、更優選80-100℃的溫度下,以及在優選10托以下、更優選5托以下、更加優選1托以下的真空度的壓力下進行優選10-74小時、更優選10-48小時、更加優選24-48小時。在這種條件下通過升華進行純化可以使共聚物的分子量變化最小化并從共聚物中除去雜質。本發明實施方案的藥物組合物包含選自紫杉醇和多西紫杉醇的一種或多種水溶性差的藥物作為活性成分。根據本發明的一個實施方案,藥物組合物還可包含作為另外的活性成分的除紫杉醇和多西紫杉醇之外的一種或多種水溶性差的藥物。作為這樣的另外的活性成分,可以使用一種或多種選自7-表紫杉醇、t-乙酰基紫杉醇、10-去乙酰基紫杉醇、10-去乙酰基-7-表紫杉醇、7-木糖基紫杉醇、10-去乙酰基-7-戊二酰紫杉醇、7-N,N-二甲基甘氨酰紫杉醇、7-L-丙氨酰紫杉醇和卡巴他賽(cabazitaxel)的紫杉烷抗癌劑。本發明實施方案的藥物組合物可包含,基于100重量份的兩親性嵌段共聚物,0.1-50重量份、更具體地0.5-30重量份的水溶性差的藥物。與兩親性嵌段共聚物的量相比,如果水溶性差的藥物的量太少,則每種藥物使用的兩親性共聚物的重量比較高,因此重構時間會增加。另一方面,如果水溶性差的藥物的量太多,則可能存在水溶性差的藥物快速沉淀的問題。如本文所用的水溶性差的藥物的“初始”量是指制備藥物組合物時摻入的水溶性差的藥物的重量。在本發明的實施方案中,當所述藥物組合物在加速條件下(40℃)儲存6個月時,其含有,以所述水溶性差的藥物的初始量為100重量份計,0.2重量份以下的由下式1表示的相關化合物:[式1]其中:R1為H或COCH3,R2為苯基或O(CH3)3。根據本發明的一個實施方案,水溶性差的藥物為紫杉醇,相關化合物可包括下式1a表示的化合物:[式1a]當在加速條件下(40℃)儲存6個月時,本發明實施方案的藥物組合物可含有,以所述水溶性差的藥物的初始量為100重量份計,0.2重量份以下、優選0.18重量份以下、更優選0.16重量份以下、更加優選0.13重量份以下、最優選0.1重量份以下的式1(特別是式1a)的相關化合物。在本發明的優選實施方案中,當在加速條件下(40℃)儲存6個月時,藥物組合物可含有,以所述水溶性差的藥物的初始量為100重量份計,小于0.15重量份、尤其小于0.10重量份以下的式1(特別是式1a)的相關化合物。當在嚴格條件下(80℃)儲存3周時,本發明實施方案的藥物組合物可含有,以所述水溶性差的藥物的初始量為100重量份計,1.2重量份以下、優選0.9重量份以下、更優選0.7重量份以下、更加優選0.4重量份以下、最優選0.2重量份以下的式1(特別是式1a)的相關化合物。在本發明的優選實施方案中,當在嚴格條件下(80℃)儲存3周時,藥物組合物可含有,以所述水溶性差的藥物的初始量為100重量份計,小于1.12重量份的式1(特別是式1a)的相關化合物。在本發明的實施方案中,含有規定限量以內的量的特定相關化合物的藥物組合物是商業可獲得的組合物,因為其可以大規模生產。在一個實施方案中,本發明的藥物組合物完全不含醚,例如乙醚。在一個實施方案中,本發明的藥物組合物完全不具有金屬鹽,例如堿金屬鹽和/或用于鹽析的鹽,例如NaCl或KCl。本發明實施方案的藥物組合物可以通過包括以下步驟的方法制備:(a)純化包含親水性嵌段(A)和疏水性嵌段(B)的兩親性嵌段共聚物;(b)將選自紫杉醇和多西紫杉醇的一種或多種水溶性差的藥物以及純化的兩親性嵌段共聚物溶解在有機溶劑中;和(c)向步驟(b)中獲得的溶液中加入水性溶劑以形成聚合物膠束。兩親性嵌段共聚物的純化如上所述,可用常規方法形成聚合物膠束。在制備本發明實施方案的藥物組合物的方法中,可以使用例如選自醇(例如乙醇)、丙酮、四氫呋喃、乙酸、乙腈和二氧六環及其組合的水混溶性有機溶劑作為有機溶劑,但并不限于此。另外,可以使用選自常規水、蒸餾水、注射用蒸餾水、生理鹽水、5%葡萄糖、緩沖液及它們的組合中的一種作為水性溶劑,但不限于此。制備本發明藥物組合物的方法還可包括在所述步驟(a)之后除去有機溶劑。在一個實施方案中,所述方法還可包括加入凍干助劑以凍干膠束組合物。可以為凍干組合物添加凍干助劑以維持餅狀。在另一個實施方案中,凍干助劑可以是選自糖和糖醇中的一種或多種。糖可以是選自乳糖、麥芽糖、蔗糖或海藻糖中的一種或多種。糖醇可以是選自甘露醇、山梨醇、麥芽糖醇、木糖醇和乳糖醇中的一種或多種。凍干助劑還可用于促進凍干的聚合物膠束組合物在重構時的均勻溶解。基于凍干組合物的總重量,凍干助劑的含量可以為1-90重量%,特別是1-60重量%,更特別是10-60重量%。通過以下實施例對本發明進行更詳細地說明。然而,這些實施例僅試圖說明本發明,且實施例不以任何方式對本發明的范圍進行限制。實施例制備例1:由單甲氧基聚乙二醇和D,L-丙交酯組成的二嵌段共聚物(mPEG-PDLLA)的合成和通過升華方法純化將150g單甲氧基聚乙二醇(mPEG,數均分子量=2,000)加入到配備有攪拌器的500ml圓底燒瓶中,并在120℃真空條件下攪拌2小時以除去水分。將溶解在200μl甲苯中的0.15g辛酸錫(Sn(Oct)2)加入到反應燒瓶中,并在真空條件下再攪拌1小時以蒸餾并除去甲苯。然后加入150gD,L-丙交酯并在氮氣氛下攪拌以溶解。在D,L-丙交酯完全溶解之后,將反應器緊密密封,并在120℃下進行聚合反應10小時。反應終止后,在磁棒攪拌下,將反應器連接到真空泵,產物在1托或更低的壓力下通過升華方法純化7小時,得到262g熔融狀態的mPEG-PDLLA。通過用1H-NMR分析獲得關于單甲氧基聚乙二醇的末端基團-OCH3的適當峰的相對強度,從而計算分子量(Mn:~3740)。制備例2:通過升華方法純化二嵌段共聚物(mPEG-PDLLA)將在制備例1的聚合反應過程中獲得的進行純化過程之前的30gmPEG-PDLLA加入單頸燒瓶中并在80℃下溶解。在磁棒攪拌下,將反應器連接到真空泵,并且在1托或更低的壓力下通過升華方法純化產物24小時和48小時。制備例3:通過升華方法純化二嵌段共聚物(mPEG-PDLLA)除了純化溫度為100℃以外,通過與制備例2相同的方法進行純化。制備例4:通過升華方法純化二嵌段共聚物(mPEG-PDLLA)除了純化溫度為120℃以外,通過與制備例2相同的方法進行純化。制備例5:使用氧化鋁(Al2O3)通過吸附法純化二嵌段共聚物(mPEG-PDLLA)將在制備例1的聚合反應過程中獲得的進行純化過程之前的30gmPEG-PDLLA加入單頸燒瓶中并加入丙酮(60ml)溶解。向其中加入氧化鋁(15g)并完全混合。將單頸燒瓶與旋轉蒸發器連接,內容物在50℃、60rpm下混合2小時。然后將溶液在室溫下用PTFE濾紙(1μm)過濾以除去氧化鋁。使用旋轉蒸發器在60℃真空下蒸餾經過濾的丙酮溶液以除去丙酮,由此獲得純化的mPEG-PDLLA。通過用1H-NMR分析獲得關于單甲氧基聚乙二醇的末端基團-OCH3的適當峰的相對強度,從而計算分子量(Mn:~3690)。根據上述制備例2-5中的純化條件的mPEG-PDLLA的分子量變化示于下表1中。表1從表1的結果可以看出,隨著純化溫度變高,mPEG-PDLLA的分子量的減少量增加。80-100℃和24-48小時,特別是100℃和24小時的純化條件可被認為是有效的。實施例1:含有紫杉醇的聚合物膠束組合物的制備稱量1g紫杉醇和5g制備例1中得到的mPEG-PDLLA,加入4ml乙醇,在60℃下攪拌直至混合物完全溶解形成澄清溶液。然后使用配備有圓底燒瓶的旋轉蒸發器在60℃下減壓蒸餾3小時以除去乙醇。然后將溫度降低至50℃,加入140ml室溫下的蒸餾水,并進行反應直至溶液變成澄清的藍色以形成聚合物膠束。向其中加入2.5g無水乳糖作為凍干助劑并使其完全溶解,使用孔徑為200nm的過濾器過濾,并冷凍干燥,得到含有紫杉醇的粉末狀聚合物膠束組合物。實施例2:含有紫杉醇的聚合物膠束組合物的制備除了使用在制備例3中純化24小時的mPEG-PDLLA之外,通過與實施例1相同的方法制備含有紫杉醇的聚合物膠束組合物。實施例3:含有紫杉醇的聚合物膠束組合物的制備除了使用在制備例5中純化的mPEG-PDLLA以外,通過與實施例1相同的方法制備含有紫杉醇的聚合物膠束組合物。實驗例1:通過液相色譜分離相關化合物向容納有已經過6個月加速試驗(溫度:40℃)的100mg含有紫杉醇的聚合物膠束組合物的小瓶中加入16.7ml去離子水(DW),使內容物完全溶解,取總量的液體并轉移到20ml容量瓶中,稀釋至標線使總體積為20ml(5.0mg/ml)。取2ml該液體并轉移到10ml容量瓶中,加入乙腈至標線使總體積為10ml(1mg/ml)。對于上述組合物,使用以下液相色譜法分離相關化合物并分段收集。液相色譜條件1)柱:Poroshell120PFP(4.6×150mm,2.7μm,Agilent)2)流動相:A:DW/B:乙腈3)流速:0.6ml/分鐘4)注射量:10μl5)檢測器:紫外吸收分光光度計(測量波長:227nm)得到的HPLC分析的色譜圖顯示在圖1中。實驗例2:紫杉醇的熱分解試驗在實驗例1中從含有紫杉醇的聚合物納米顆粒組合物中分段收集的相關化合物中,許多聚合物同時存在,因此很難進行直接實驗。作為使用LC/MS/MS進行的初步實驗中的定性分析結果,相關化合物被認為是由紫杉醇與水結合產生的化合物。因此,作為加入水分子的方法,進行加熱紫杉醇的實驗以確認是否產生推測的相關化合物。首先,將1g紫杉醇在170℃下保持2~3小時,并完全溶解在45ml乙腈中,然后向其中加入5mlDW。通過使用該溶液,在制備型LC上分離RRT0.96的相關化合物并分段收集。實驗例3:使用LC/MS/MS對相關化合物進行定性分析通過液相色譜和質譜儀(LC/MS/MS)對在實驗例1-2中分離的相關化合物(RRT:0.96±0.02(0.94~0.98))進行分析。根據HPLC分析結果,在實驗例2中分段收集的物質與聚合物膠束組合物(圖4)中的RRT0.96處的相關化合物在相同的位置處顯示HPLC峰。通過LC/MS/MS對該物質進行進一步的分析。首先作為MS掃描的結果,兩種物質均顯示了m/z894.1amu,其為[M+H2O+Na]+(圖2和圖3)。然后進行產物離子掃描,其結果示于圖4中。一起顯示的還有經過六個月加速試驗的含有紫杉醇的聚合物納米顆粒組合物中形成的RRT0.96的相關化合物的結果。總之,可以證實在實驗例2中熱分解紫杉醇之后在RRT0.96處分段收集的物質是與聚合物膠束組合物經過六個月加速試驗之后產生的在RRT0.96位置處的相關化合物具有相同結構的化合物。在下面的測量中,使用液相色譜1200系列和電噴霧電離質譜儀6400系列(Agilent,美國)作為LC/MS/MS。分析條件如下。液相色譜條件1)柱:Poroshell120PFP(4.6×150mm,2.7μm,Agilent)2)流動相:A:DW/B:乙腈時間(分鐘)A%B%0.00653525.00455528.00455530.00653535.0065353)流速:0.6ml/分鐘4)注射量:10μl5)檢測器:紫外吸收分光光度計(測量波長:227nm)電噴霧電離質譜儀的條件1)電離:電噴霧電離,正(ESI+)2)MS方法:MS2掃描/產物離子掃描3)離子源:AgilentJetStreamESI4)霧化器氣體(壓力):氮氣(35psi)5)離子噴霧電壓:3500V6)干燥氣體溫度(流速):350℃(7L/分鐘)7)鞘氣溫度(流速):400℃(10L/分鐘)8)碎裂電壓:135V9)噴嘴電壓:500V10)池加速電壓:7V11)EMV:0V12)碰撞能量:22V13)前體離子:m/z836.214)質量掃描范圍:m/z100~1500將從檢測階段分離并產生的用于分析的物質設置為在質譜儀中流動,同時定性分析相關化合物的檢測離子,并選擇質譜的特征離子[M+Na]。實驗例4:對由通過熱分解紫杉醇所得混合物在RRT0.96處獲得的物質進行NMR分析通過NMR對實驗例2中由通過熱分解紫杉醇所得混合物在RRT0.96處獲得的物質進行分析。在NMR分析中,1HNMR分析的結果顯示在圖5中,13CNMR分析的結果顯示在圖6中,COSY(相關光譜)分析的結果顯示在圖7中,HMBC(異核多鍵相關光譜)分析示于圖8中。根據分析結果,可以確認由通過熱分解紫杉醇所得混合物在RRT0.96處獲得的物質(即經過六個月加速試驗的本發明的含有紫杉醇的聚合物膠束組合物中的相關化合物(RRT:0.96±0.02(0.94~0.98))是以下的紫杉醇與水的結合形式。紫杉醇與一分子水的結合形式:C47H53NO15(871.94g/mol)。核磁共振光譜的條件1.1H1)NMR設備:裝備有溫度控制器的BruckerDRX-9002)樣品/溶劑:在外徑為5mm的NMR管中的1-10mg樣品/0.6mL氯仿-d(在所有NMR實驗中,使用相同的樣品)3)探頭:Brucker5mmCPTCI4)質子90°脈沖寬度/激發角/捕獲時間:7.4微秒/30°/3.3秒5)弛豫延遲/掃描次數:2.0秒/162.13C1)探頭:Brucker5mmCPTCI2)碳90°脈沖寬度/激發角/捕獲時間:11.8微秒/30°/0.58秒3)弛豫延遲/掃描次數:3.0秒/6563.COSY1)NMR設備:BruckerDRX-9002)探頭:Brucker5mmCPTCI3)脈沖序列:cosygpqf脈沖序列4)質子90°脈沖寬度/捕獲時間:7.4微秒/0.13秒5)弛豫延遲/掃描次數/ω1的實驗次數:1.5秒/4/3204.HMBC1)NMR設備:BruckerDRX-9002)探頭:Brucker5mmCPTCI3)脈沖序列:hmbcgplpndqf脈沖序列4)質子90°脈沖寬度/碳90°脈沖寬度/捕獲時間:7.7微秒/11.8微秒/0.12秒5)弛豫延遲/掃描次數/ω1的實驗次數:1.5秒/4/3206)溫度/1/2(JCH):283K/3.5毫秒。實驗例5:在嚴格條件下(80℃)含有藥物的聚合物膠束的儲存穩定性的對比試驗將在實施例1-3中制備的紫杉醇的聚合物膠束組合物在80℃的烘箱中保持3周,然后用HPLC分析組合物以比較相關化合物的量。通過將膠束組合物溶解在80%乙腈水溶液中并稀釋至紫杉醇濃度為600ppm來制備測試溶液。HPLC分析所得的色譜圖顯示在圖9中,并且按照嚴格試驗時間的相關化合物的含量變化(%)示于下表2中。HPLC條件柱:直徑2.7μm,Poroshell120PFP(4.6×150mm,2.7μm)(Agilent柱)流動相時間(分鐘)水:乙腈0~2565:35→45:5525~2845:5528~3045:55→65:3530~3565:35檢測器:紫外吸收分光光度計(227nm)流速:0.6ml/分鐘各相關化合物的量(%)=100(Ri/Ru)Ri:在測試溶液分析中檢測到的每種相關化合物的面積Ru:在測試溶液分析中檢測到的所有峰面積的總和[表2]*RRT0.87±0.02:紫杉醇,氧雜環丁烷開環化合物RRT0.96±0.02:紫杉醇,氧雜環丁烷開環化合物RRT1.00:紫杉醇RRT1.10±0.02:紫杉醇,L-丙交酯反應化合物RRT1.12±0.02:紫杉醇,D-丙交酯反應化合物RRT1.44±0.05:紫杉醇,去水化合物從表2和圖9可知,與實施例1的組合物相比,實施例2或3的聚合物膠束藥物組合物的穩定性得到提高,紫杉醇量的減少相對較小,因此可以更穩定地維持組合物中所含藥物的效果。實驗例6:在加速條件下(40℃)含有藥物的聚合物膠束的儲存穩定性的對比試驗除了將在實施例1中制備的紫杉醇的聚合物膠束組合物在穩定性試驗儀中于40℃下保持6個月之外,通過與實驗例5相同的方法進行實驗。將按照加速試驗時間的相關化合物的含量變化示于下表3中。[表3]上述試驗結果顯示了對不同批次的3種或更多種聚合物膠束組合物進行的試驗中每種相關化合物和紫杉醇的量的平均值。每個相關化合物的量在批次之間顯示出差異,下表表示在每個批次的測試中檢測到最多相關化合物的批次的情況。[表4]在藥物的質量控制中,由于特定雜質的最高值與其平均值是同樣重要的,因此必須改進組合物的質量以使這些值可從根本上降低。通過實驗實施例5,已經證明實施例2或3的聚合物膠束藥物組合物與實施例1的組合物相比顯示更低量的相關化合物。因此,可以推斷如果在加速儲存溫度下(40℃)儲存6個月,與上述表3中所示的實施例1的組合物的量相比,實施例2或3的組合物會具有更低量的相關化合物。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1