含偶氮苯衍生物的脂質體及其制備方法和應用與流程
本發明涉及刺激響應載體領域,尤其涉及一種含偶氮苯衍生物的脂質體及其制備方法和應用。
背景技術:
納米技術的突破對多個行業產生了重要的影響,尤其是對材料科學、生物技術和藥物治療。不同的納米載體,如脂質體、聚合物、膠束和碳基納米材料正逐漸應用到醫學領域,例如治療性藥物大分子的運輸(藥物和基因)。但是,目前治療性藥物大分子的運輸仍面臨著很大的挑戰,因為我們所需要的藥物運輸體系最好能夠實現藥物的定時、定點和定量釋放。對用于藥物運輸體系,目前已經有一定的研究,例如:通過智能化微米/納米粒子(MNPs)來實現。所述MNPs是指能夠以某種具體的方式響應外部/內部刺激,進而實現內部藥物的可控釋放的微米/納米粒子的總稱。光照由于其強度、波長、照射時間等參數可以精確調節,并且對生物體是非侵入性的,所以,能夠對光刺激進行響應的MNPs具有廣闊的應用前景。
因此,需要一種能夠對光刺激進行響應的含偶氮苯衍生物的脂質體。
技術實現要素:
本發明的目的在于提供了一種含偶氮苯衍生物的脂質體及其制備方法和應用,所述含偶氮苯衍生物的脂質體能夠對光刺激進行響應。
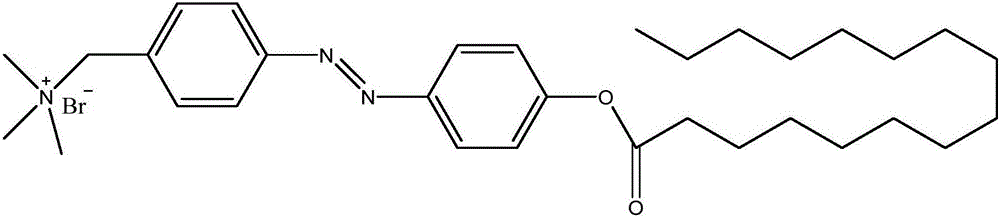
為實現上述目的,本發明提供了一種含偶氮苯衍生物的脂質體,所述含偶氮苯衍生物的脂質體中的偶氮苯衍生物、磷脂和膽固醇的摩爾比為:1~2:1~2:1~2;
所述偶氮苯衍生物的結構式如下:
其中,R1為C1~6的直鏈烷基;R2為C12~16的直鏈疏水性烷基;X為:O或NH。
其中,所述含偶氮苯衍生物的脂質體還包括:包封在所述含偶氮苯衍生物的脂質體內的藥物。
其中,所述藥物為阿霉素。
為了進一步實現發明目的,本發明還提供了一種含偶氮苯衍生物的脂質體的制備方法,所述偶氮苯衍生物的結構式如下:
其中,R1為C1~6的直鏈烷基;R2為C12~16的直鏈疏水性烷基;X為:O或NH;
所述含偶氮苯衍生物的脂質體的制備方法包括以下步驟:
A、將摩爾比為:1~2:1~2:1~2的偶氮苯衍生物、磷脂和膽固醇溶于有機溶劑中,旋干,得脂質薄膜;
B、加入水化試劑,然后進行超聲處理,得含偶氮苯衍生物的脂質體。
其中,所述有機溶劑為按2:1體積比配置的氯仿和甲醇混合液。
其中,所述水化試劑為pH=7.4的檸檬酸溶液或pH=7.4的磷酸緩沖液。
其中,所述步驟B具體為:
在瓶壁上有脂質薄膜的瓶中加入適量水化試劑,用探頭式超聲波細胞破碎儀超聲10~40min,然后靜置于25~42℃環境中,得含偶氮苯衍生物的脂質體懸液。
其中,所述含偶氮苯衍生物的脂質體的制備方法還包括以下步驟:
C、利用pH梯度法,將阿霉素包封在含偶氮苯衍生物的脂質體中,得內包阿霉素的含偶氮苯衍生物的脂質體。
其中,所述步驟C具體為:
將阿霉素加入至含偶氮苯衍生物的脂質體懸液中,充分溶解,然后用NaOH調pH至7.4,8 000~12 000g離心,所得上層液體即為內包阿霉素的含偶氮苯衍生物的脂質體。
為了進一步實現本發明的目的,本發明還提供了上述含偶氮苯衍生物的脂質體在制備藥物過程中的應用。
本發明的含偶氮苯衍生物的脂質體能夠對紫外光進行響應,當所述含偶氮苯衍生物的脂質體包封有藥物時,可通過紫外線的照射來實現該含偶氮苯衍生物的脂質體內部藥物的可控釋放,從而減輕藥物的副作用,提高治療效果。本發明的含偶氮苯衍生物的脂質體的制備方法簡單,效率高。
附圖說明
圖1是本發明第一具體制備實施例制備所得偶氮苯衍生物的結構式。
圖2是本發明第二具體制備實施例制備所得偶氮苯衍生物的結構式。
圖3是本發明第三具體制備實施例制備所得偶氮苯衍生物的結構式。
圖4是本發明第三具體制備實施例制備所得偶氮苯衍生物在經紫外光照射后的紫外-可見吸收光譜變化圖。
圖5是本發明第四具體制備實施例制備所得內包阿霉素的含偶氮苯衍生物的脂質體的光控釋放曲線。
具體實施方式
下面結合具體實施例對本發明做進一步的詳細說明,以下實施例是對本發明的解釋,本發明并不局限于以下實施例。
在本發明的第一實施例中,一種偶氮苯衍生物,結構式如下圖1所示:
其中,R1為C1~6的直鏈烷基;R2為C12~16的直鏈疏水性烷基;X為:O或NH。
本發明的偶氮苯衍生物的一端的直鏈疏水性烷基鏈能夠插入脂質體的磷脂雙分子層中,另一端的親水性陽離子季銨鹽基團則能朝向磷脂雙分子層膜外的水溶液,從而在脂質體中形成規律性的排布。本發明的偶氮苯衍生物在紫外光照射下可以發生可逆的構象改變,在未經紫外線照射時,保持反式構型,經紫外線照射后,變換成順式構型。
優選的,所述R1為C2~4的直鏈烷基,即,乙烷基、丙烷基或正丁烷基。
優選的,所述R2為C12、C14或C16的直鏈疏水性烷基。
在本發明的第二實施例中,一種制備上述偶氮苯衍生物的方法,包括以下步驟:
A、使化合物1和化合物2反應,得化合物3;
B、將化合物3和N-溴代琥珀酰亞胺在惰性氣體氛圍下溶于四氯甲烷并在過氧化苯甲酰的催化下發生溴化反應,得化合物4;
C、使化合物4與第一底物反應,得所述偶氮苯衍生物;
所述化合物1的結構式如下:
所述化合物2的結構式如下:
X2-R2;
所述化合物3的結構式如下:
所述化合物4的結構式如下:
所述X1為OH或NH2;
所述X2為與X1反應形成X的COOH或COCl;
所述第一底物的結構式為:
需要說明的是,所述惰性氣體包括氦氣、氬氣、氖氣或氙氣中的至少一種。
在本發明的一具體實施例中,所述X1為NH2;所述X2為COCl;所述步驟A包括以下步驟:
A1、將化合物1溶于二氯甲烷,然后加入三乙胺,得第一反應溶液;
A2、將化合物2溶于二氯甲烷,得第二反應溶液;
A3、將第二反應溶液滴入第一反應溶液中,常溫攪拌過夜;
A4、除去步驟A3的產物中的二氯甲烷,得固體;
A5、將固體溶于二氯甲烷,并通過柱層析進行分離純化,所述流動相為按1:1體積比配制的石油醚和二氯甲烷混合液,得化合物3。
需要說明的是,在所述步驟A中,二氯甲烷主要作為反應溶劑,三丁胺為催化劑;步驟A4可通過真空干燥或旋蒸實現。另外,在本方案中,化合物1中的NH2和化合物2中的COCl反應,從而生成化合物3,這種方法與通過化合物中的OH或NH2和化合物2中的COOH反應生成化合物3的方法相比,反應效率更高,制備得到化合物3的得率更高。
在上一具體實施例的基礎上,本發明提出另一具體實施例,所述步驟A還包括以下步驟:
A0、將正十二烷酸加入氯化亞砜,加熱至60-70℃,攪拌過夜,進而除去未反應的氯化亞砜,得化合物2。
需要說明的是,步驟A0中可通過真空干燥或旋蒸等方法除去氯化亞砜。
在本發明的另一具體實施例中,所述步驟B包括以下步驟:
B1、將化合物3、N-溴代琥珀酰亞胺和過氧化苯甲酰在氬氣氛圍下溶于四氯甲烷,加熱回流反應12-72小時;
B2、將反應體系冷卻至0℃,抽濾得粉末;
B3、用乙醚洗滌粉末,得化合物4。
需要說明的是,步驟B1中的過氧化苯甲酰為催化劑;步驟B2抽濾所得粉末為化合物4的粗產物;步驟B3中用乙醚洗滌粉末,主要是通過乙醚將未反應的雜質去除,從而起到純化的作用。
在本發明的另一具體實施例中,所述步驟C包括以下步驟:
C1、將化合物4溶于乙醇,再加入第一底物,回流反應6-48小時;
C2、將步驟C1的產物蒸干,再用按5:1體積比配制的乙醚和二氯甲烷混合液溶解,過濾收集固體沉淀。
需要說明的是,步驟C2中,可通過旋蒸將步驟C1的產物蒸干;步驟C2所得固體沉淀為本發明所述偶氮苯衍生物的粗產物。
基于上一具體實施例,本發明提出另一具體實施例,所述步驟C還包括以下步驟:
C3、用按5:1體積比配制的乙醚和二氯甲烷混合液洗滌固體沉淀,得所述偶氮苯衍生物。
需要說明的是,所述按5:1體積比配制的乙醚和二氯甲烷混合液能夠溶解未發生反應的雜質,從而起到提純的作用。
在本發明的第一具體制備實施例中,所述偶氮苯衍生物的結構式如圖1所示,該偶氮苯衍生物的制備方法包括以下步驟:
S01、使化合物1(4-甲基-4’羥基偶氮苯)與化合物2(正十四烷酸)在濃硫酸的催化和加熱條件下發生脂化反應,生成化合物3(4-甲基-4’正十四烷酸偶氮苯酯);
S02、將4-甲基-4’正十四烷酸偶氮苯酯、N-溴代琥珀酰亞胺和過氧化苯甲酰在氬氣氛圍下溶于四氯甲烷,加熱回流反應24小時;
S03、將步驟S02的反應體系冷卻至0℃,抽濾得粉末;
S04、用乙醚洗滌步驟S03所得粉末,除去未反應的雜質,得化合物4(4-溴甲基-4’正十四烷酸偶氮苯酯);
S05、將4-溴甲基-4’正十四烷酸偶氮苯酯溶于乙醇,然后加入三乙胺,回流反應12小時;
S06、將溶液蒸干,再用按5:1體積比配制的乙醚和二氯甲烷混合液溶解,過濾收集固體沉淀,即得圖1所示偶氮苯衍生物,即4-(N,N,N-三乙胺甲基溴化物)-4’-正十四烷酸偶氮苯酯。
在本發明的第二具體制備實施例中,所述偶氮苯衍生物的結構式如圖2所示,該偶氮苯衍生物的制備方法包括以下步驟:
S01、使化合物1(4-甲基-4’羥基偶氮苯)與化合物2(正十六烷酸)在濃硫酸的催化以及加熱條件下發生脂化反應,生成化合物3(4-甲基-4’-正十六烷酸偶氮苯酯);
S02、將4-甲基-4’-正十六烷酸偶氮苯酯、N-溴代琥珀酰亞胺和過氧化苯甲酰在氬氣氛圍下溶于四氯甲烷,加熱回流反應48小時;
S03、將步驟S02的反應體系冷卻至0℃,抽濾得粉末;
S04、用乙醚洗滌步驟S03所得粉末,除去未反應的雜質,得化合物4(4-溴甲基-4’-正十六烷酸偶氮苯酯);
S05、將4-溴甲基-4’-正十六烷酸偶氮苯酯溶于乙醇,然后加入三甲胺,回流反應24小時;
S06、將溶液蒸干,再用按5:1體積比配制的乙醚和二氯甲烷混合液溶解,過濾收集固體沉淀,即粗產物。
S07、用按5:1體積比配制的乙醚和二氯甲烷混合液洗滌固體沉淀3次,得圖2所示偶氮苯衍生物,即4-(N,N,N-三甲胺甲基溴化物)-4’-正十六烷酸偶氮苯酯。
在本發明的第三具體制備實施例中,所述偶氮苯衍生物的結構式如圖3所示,該偶氮苯衍生物的制備方法包括以下步驟:
S01、將2g正十二烷酸加入10mL氯化亞砜中,65℃攪拌過夜,旋蒸出去未反應得氯化亞砜,得到2g無色油狀粗產物,即化合物2(正十二酰氯);
S02、將0.62g化合物1(4-甲基-4’氨基偶氮苯)溶于20mL二氯甲烷,然后加入0.59g三乙胺,得第一反應溶液;
S03、將0.97g無色油狀粗產物(正十二酰氯)溶于5mL二氯甲烷,得第二反應溶液;
S04、將第二反應溶液慢慢滴入第一反應溶液中,25℃攪拌過夜;
S05、真空干燥除去步驟S04的產物中的二氯甲烷,得黃色固體;
S06、用二氯甲烷溶解步驟S05所得黃色固體,同時加入200-300目的硅膠攪拌,旋蒸除去二氯甲烷后上樣,加流動相進行柱層析,所述流動相為按1:1體積比配制的石油醚和二氯甲烷混合液;得純化后的化合物3(4-甲基-4’正十二酰氨基偶氮苯)1.12g;
S07、將0.5g 4-甲基-4’正十二酰氨基偶氮苯、0.34g N-溴代琥珀酰亞胺和15mg過氧化苯甲酰在氬氣氛圍下溶于四氯甲烷,加熱回流反應48小時;
S08、將步驟S07產物冷卻至0℃,抽濾得黃色粉末;
S09、用乙醚洗滌黃色粉末,除去未反應的雜質,得化合物4(4-溴甲基-4’-正十二酰氨基偶氮苯);
S10、將250mg 4-溴甲基-4’-正十二酰氨基偶氮苯溶于6mL乙醇,然后加入1mL三丁胺和2mL乙醇,回流反應24小時;
S11、將步驟S10的產物蒸干,再用20mL按5:1體積比配制的乙醚和二氯甲烷混合液溶解,過濾收集橘黃色固體沉淀;
S12、用20mL按5:1體積比配制的乙醚和二氯甲烷混合液洗滌橘黃色固體沉淀三次,得120mg圖3所示偶氮苯衍生物,即4-(N,N,N-三丁胺甲基溴化物)-4’-正十二酰氨基偶氮苯。
為了進一步證明本發明的偶氮苯衍生物的光響應特性,本發明的發明人設計了以下試驗,以氯仿或乙醇為溶劑,將所述偶氮苯衍生物配置成約1μmol/L的溶液,并測定其紫外可見吸收光譜,然后用紫外光照射不同的時間,測各個時間點的吸收光譜,然后按不同的紫外光照射時間,分別作吸光度和波長的曲線。其中,以第三具體實施例所得偶氮苯衍生物的結果如圖4所示。如圖4所示,隨著紫外線照射時間的延長,該偶氮苯衍生物在350nm處的吸光度值不斷降低,說明該偶氮苯衍生物可以發生快速的反式-順式異構,可作為一種光控開關。
需要說明的是,本發明的其他偶氮苯衍生物,例如上述第一具體制備實施例、第二具體制備實施例中所得的偶氮苯衍生物,在上述試驗中的結果與圖4類似(圖未示),說明本發明的偶氮苯衍生物可以發生快速的反式-順式異構,可作為一種光控開關。
在本發明的第三實施例中,一種制備含上述偶氮苯衍生物的脂質體的方法,包括以下步驟:
A、將摩爾比為:1~2:1~2:1~2的偶氮苯衍生物、磷脂和膽固醇溶于有機溶劑中,旋干,得脂質薄膜;
B、加入水化試劑,然后進行超聲處理,得含偶氮苯衍生物的脂質體。
需要說明的是,在第三實施例中,所述磷脂為磷脂酰膽堿、磷脂酰乙醇胺、磷脂酸、磷脂酰絲氨酸、磷脂酰甘油、磷脂酰肌醇、或它們的任意組合物。所述機溶劑包括氯仿、二氯甲烷、甲醇、乙醇和乙醚中的至少一種。所述水化試劑用于給含偶氮苯衍生物的脂質體提供穩定的環境,形成脂質體懸液。
第三實施例中通過超聲分散法制備含偶氮苯衍生物的脂質體,制備方法簡單,效率高。
若要制備包封有藥物的含上述偶氮苯衍生物的脂質體,根據藥物性質的不同,可在第三實施例的方法的基礎上,通過不同的處理獲得。
在本發明的一個實施例中,當藥物為水溶性藥物時,可將適量的藥物溶于活性溶液中,例如pH=7.4的磷酸鹽緩沖液(PBS),然后將該溶液加入瓶壁上有脂質薄膜的瓶中,用探頭式超聲波細胞破碎儀超聲10~40min,然后靜置于25~42℃環境中靜置1~6小時。將所得產物加入透析袋中,并在pH=7.4的磷酸鹽緩沖液(PBS)中透析,除去未被包封的藥物,最終獲得內包藥物的含偶氮苯衍生物的脂質體懸液。
需要說明的是,本實施例中,所述溶有藥物的活性溶液的量,可根據瓶中瓶壁上的脂質薄膜的覆蓋范圍進行調整;所述透析袋的分子量大小根據藥物的分子量進行選擇。
在本發明的另一個實施例中,當藥物為脂溶性藥物,例如阿霉素時,可將適量的300mM的檸檬酸溶液(pH=7.4)加入瓶壁上有脂質薄膜的瓶中,用探頭式超聲波細胞破碎儀超聲10~40min,然后靜置于25~42℃環境中靜置1~6小時。然后用pH梯度法包封阿霉素(DOX),獲得內包阿霉素的含偶氮苯衍生物的脂質體懸液。
需要說明的是,本發明所述的內包藥物可為水溶性藥物,例如:鹽酸吉西他濱、卡鉑、鹽酸阿糖胞苷、鹽酸氮芥、鹽酸米托蒽醌;也可為脂溶性藥物,例如阿霉素、紫杉醇等。
在本發明的第四具體制備實施例中,所述偶氮苯衍生物為上述第三具體制備實施例制備所得的產物;所述藥物為阿霉素。
1)將磷脂、膽固醇、偶氮苯衍生物按照1:1:1的摩爾比(各0.02mmol)一起溶于按2:1體積比配置的20mL氯仿和甲醇混合液,充分溶解后,在37℃恒溫水浴下用旋轉蒸發器除去有機溶劑,圓底燒瓶底形成一層均勻脂質薄膜。
2)然后加入10mL,300mM的檸檬酸溶液(pH=7.4),將脂質薄膜水化下來,接著用探頭式超聲波細胞破碎儀超聲30min(功率100mW,on 30s,off 30s),然后置于37℃水浴中放置2小時以完成封閉過程,所得即為脂質體懸液。
3)用pH梯度法包封阿霉素(DOX):稱取2mg DOX,加入到脂質體懸液中,充分溶解,用1.0M NaOH調pH至7.4。將脂質體溶液10 000g離心10min,除去超聲過程中探頭上掉下來的鈦顆粒以及未分散的脂質,上層液體即為內包阿霉素的含偶氮苯衍生物的脂質體懸液。
為了驗證所得內包阿霉素的光響應脂質體的光控釋放效果,本發明的發明人設計了另外一個驗證試驗。
實驗組:將第四具體制備實施例制備的內包阿霉素的含偶氮苯衍生物的脂質體懸液5mL分散在45mL pH=7.4的PBS緩沖液中,然后將所得的溶液放置在黑暗環境中,每小時用紫外光照射10min,每2個小時測定一次PBS緩沖液中被釋放出的阿霉素含量。
對照組:將第四具體制備實施例制備的內包阿霉素的光響應脂質體5mL分散在45mL pH=7.4的PBS緩沖液中,然后將所得的溶液放置在黑暗環境中,同樣每2個小時測定一次PBS緩沖液中被釋放出的阿霉素含量。
實驗組、對照組各做3個重復。
以每個時間點PBS緩沖液中被釋放出的阿霉素含量與該PBS緩沖液中的總阿霉素量的比值為縱坐標,時間為橫坐標,作曲線,結果如圖5所示。
如圖5所示,在紫外線光照下,內包阿霉素的光響應脂質體的磷脂雙分子層中的偶氮苯化合物發生反式-順式異構,對磷脂膜造成擾動,并在磷脂雙分子層中形成通道,進而使得阿霉素被釋放出,加速內包阿霉素的光響應脂質體內部的阿霉素的釋放。
因此,本發明的偶氮苯化合物能夠對光刺激進行響應,基于本發明的偶氮苯化合物制備的含偶氮苯衍生物的脂質體可制備成內包藥物的MNPs,具有廣闊的應用前景。
以上僅為本發明的優選實施例,并非因此限制本發明的專利范圍,凡是利用本發明說明書及附圖內容所作的等效結構或等效流程變換,或直接或間接運用在其他相關的技術領域,均同理包括在本發明的專利保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!