一種可注射的腫瘤靶向性熱敏前藥及其制備方法與應用與流程
本發明涉及一種可注射的腫瘤靶向性熱敏前藥及其制備方法與應用,屬于生物醫藥技術領域。
背景技術:
癌癥嚴重危害著人類的生命健康,被認為是僅次于心腦血管的致人死亡的主要原因(China Cancer,2009,18,88-89)。在中國,癌癥已成為疾病死因之首,發病率和死亡率還在攀升,癌癥已成為非常重要的公共健康問題。由于難以早期發現,其治療一直是醫學領域的難點。目前廣泛使用的許多化療藥物都存在難溶于水及在水中穩定性差等缺點,且化療藥物在抑制和殺傷腫瘤細胞的同時,對正常細胞同樣有殺傷作用,尤其是對增殖較為迅速的正常細胞,如骨髓造血細胞和胃腸道粘膜上皮細胞等。這是目前影響用藥劑量、治療效果和患者生活質量的嚴重問題(Ann.Surg.,1999,229,790-800)。因此,迫切需要開發新的治療癌癥的方式。
技術實現要素:
本發明的目的之一在于提供一種可注射的腫瘤靶向性熱敏前藥,該前藥應用于熱化療,配合腫瘤熱療可有效抑制腫瘤的生長,為癌癥的治療提供了一種新的方式。
本發明的另一目的在于提供所述可注射的腫瘤靶向性熱敏前藥的制備方法。
本發明的再一目的在于提供含所述可注射的腫瘤靶向性熱敏前藥的藥物組合物。
本發明的再一目的在于提供所述可注射的腫瘤靶向性熱敏前藥及含其的組合物的應用。
為此,本發明一方面提供一種可注射的腫瘤靶向性熱敏前藥(馬來酸酐‐藥物鍵合物),其中,該前藥由以下通式描述:
X-Y-Z
X為
Z為含酮羰基或羧基的抗腫瘤藥物或其鹽;
Y為其中A為-(CH2)m-NH-,B為含兩個以上羧基的且含偶氮基團的馬來酸酐-藥物間隔物,C為-NH-N=或-NH-NH-;
X中的N與A中-(CH2)m-相連;
B中一羧基通過酰胺鍵與A中-NH-相連;
B中另外的羧基通過酰胺鍵與C中-NH-相連;
C中-N=亞胺鍵由Z中的酮羰基縮合形成或C中-NH-通過酰胺鍵與Z中的羧基相連;
m選自0~3之間的整數。
優選地,m為1、2或3;更優選地,m為2。
優選地,B為含兩個羧基的且含偶氮基團的馬來酸酐-藥物間隔物,B中一羧基通過酰胺鍵與A中-NH-相連;B中另一羧基通過酰胺鍵與C中-NH-相連。
本發明所述腫瘤靶向性熱敏前藥應用于熱療與化療結合(即熱化療)抗腫瘤領域,其有效克服了現有技術中化療藥物難溶于水、在水中穩定性差及毒性大的缺陷。
本發明熱敏前藥具有X片段(為馬來酸酐片段),其能與白蛋白特異性結合,當將本發明熱敏前藥注射在體內后,其能與體內的白蛋白特異性結合,并靶向(腫瘤組織細胞膜表面富含gp60、gp30、gp18等白蛋白受體結合)蓄積到腫瘤部位,在腫瘤部位的給予一定的溫度刺激下可從白蛋白釋放并發揮藥效殺死腫瘤。此類熱響應機制可使該熱敏前藥在腫瘤部位定點釋放,大大增加藥物作用部位如細胞質和細胞核中的濃度,克服腫瘤的耐藥性,減少化療的毒副作用。該化合物具有生物相容性好的特點,其熱敏感響應性能夠實現藥物的特異性釋放,化合物發生斷裂后,藥物發揮抗腫瘤作用,白蛋白可自然降解,可降低體內載體的蓄積。本發明有助于推動熱化學治療的研究與發展,同時為癌癥的臨床診斷和治療提供新理論和新方法。
實驗研究表明本發明所述腫瘤靶向性熱敏前藥在正常溫度及中溫條件下對正常細胞無毒,而在中溫條件下對腫瘤細胞存在明顯的毒性,動物實驗表明本發明所述腫瘤靶向性熱敏前藥結合熱療可顯著抑制腫瘤的生長。
根據本發明的具體實施方式,在本發明所述腫瘤靶向性熱敏前藥中,B包括4,4'-偶氮雙(4-氰基戊酸)或2,2'-偶氮雙(N-(2-羧基乙基)-2-甲基丙脒)(2,2'-azobis(N-(2-carboxyethyl)-2-methylpropionamidine)等。
根據本發明的具體實施方式,在本發明所述可注射的腫瘤靶向性熱敏前藥中,Z包括阿霉素、表阿霉素、甲氨蝶呤、柔紅霉素和去甲氧基柔紅霉素或它們的鹽等中的一種或多種;優選阿霉素。
優選地,本發明所述可注射的腫瘤靶向性熱敏前藥具有如下結構:
R1為
R2為H或羥基;
R3為H或-OCH3。
更優選地,所述前藥具有如下結構:
另一方面,本發明提供腫瘤靶向性熱敏前藥的制備方法,所述方法包括如下步驟:
(a)將含兩個以上羧基的且含偶氮基團的馬來酸酐-藥物間隔物溶于有機溶劑中,加入活化羧基的活性試劑及穩定活化后羧酸的活化基團穩定劑進行反應;優選地,所述馬來酸酐-藥物間隔物、所述活性試劑與縮合劑的摩爾比為0.001~10:0.001~10:0.001~10,進一步優選0.1~5:0.1~5:0.1~10;
(b)將NH2-(CH2)m-NH2加入到步驟(a)反應后的反應液中進行反應以使NH2-(CH2)m-NH2中的一氨基與所述馬來酸酐-藥物間隔物中的一羧基通過酰胺鍵相連,并提純;優選地,步驟(a)中所述馬來酸酐-藥物間隔物與所述NH2-(CH2)m-NH2的摩爾比為0.001~10:0.001~10,進一步優選0.1~10:0.1~10;
(c)將步驟(b)提純后的化合物與馬來酸酐加入乙酸中回流反應,回流過程中滴加乙酸酐,以使NH2-(CH2)m-NH2中另一氨基與馬來酸酐縮合形成酰亞胺鍵,反應后提純;優選地,步驟(a)中所述馬來酸酐-藥物間隔物與所述乙酸酐的摩爾比為0.001~10:0.001~10,進一步優選0.1~5:0.1~5;
(d)將步驟(c)所得化合物溶于有機溶劑中,并加入N-甲基嗎啡啉,再先后滴加氯甲酸異丁酯、叔丁基咔唑鹽,以使所述馬來酸酐-藥物間隔物中另外的羧基形成酰肼;優選地,步驟(a)中所述馬來酸酐-藥物間隔物、所述N-甲基嗎啡啉、所述氯甲酸異丁酯與所述叔丁基咔唑鹽的摩爾比為0.001~10:0.001~10:0.001~10:0.001~10,進一步優選0.1~5:0.1~5:0.1~5:0.1~5;
(e)以三氟乙酸鹽處理步驟(d)所得化合物以成鹽,并提純;
(f)將步驟(e)所得化合物與含酮羰基或羧基的抗腫瘤藥物中的酮羰基或羧基反應以形成亞胺鍵或酰胺鍵,后處理形成本發明所述可注射的腫瘤靶向性熱敏前藥。
步驟(a)的作用是活化馬來酸酐-藥物間隔物中的一個羧基,步驟(b)是使NH2-(CH2)m-NH2中的一氨基與所述馬來酸酐-藥物間隔物中的一羧基通過酰胺鍵相連;步驟(c)是使NH2-(CH2)m-NH2中另一氨基與馬來酸酐縮合形成酰亞胺鍵;步驟(d)是使所述馬來酸酐-藥物間隔物中另外的羧基形成酰肼;步驟(e)是將肼成鹽,步驟(f)是使肼中的氨基與抗腫瘤藥物中的酮羰基形成亞胺或羧基形成酰胺。
本發明所述方法簡便、反應條件溫和、重現性好,可以實現規模化生產,具有非常重要的實用價值。
根據本發明的具體實施方式,在本發明所述方法中,步驟(a)中所述的活化基團穩定劑包括NHS和/或硫代-NHS(Sulfo-NHS)。
根據本發明的具體實施方式,在本發明所述方法中,步驟(a)中所述的活化劑包括EDC、DCC、DIC和CDI中的一種或多種。
在步驟(a)中還可加入適量的催化劑以加快羧酸的活化,例如吡啶等。步驟(d)中優選在極性非質子性溶劑中反應,例如四氫呋喃等,所述氯甲酸異丁酯與所述叔丁基咔唑鹽優選以溶液的形式加入,例如溶解在四氫呋喃中形成的溶液。步驟(f)中還可加入適量的催化劑催化形成亞胺反應以及形成酰胺鍵的反應。步驟(a)~步驟(f)中的反應時間、反應溫度可進一步優化。步驟(a)~步驟(f)優選在惰性環境下進行。本發明所述方法可通過控制馬來酸酐、間隔物與抗腫瘤藥物的比例,反應時間、試劑的濃度、反應的條件(加熱溫度、時間),制備各種馬來酸酐結合熱敏藥物。
優選地,本發明所述方法按如下步驟進行:
(i)準確稱取0.001~10mmol含兩個以上羧基的且含偶氮基團的馬來酸酐-藥物間隔物(例如4,4'-偶氮雙(4-氰基戊酸)),溶于0.1~100mL二甲基亞砜,再加入0.001~10mmol NHS和0.001~10mmol EDC及1~1000μL吡啶,氮氣保護下室溫反應0.5~48h;
(ii)將0.001~10mmol NH2-(CH2)m-NH2(例如乙二胺)加入步驟(i)的反應溶液中,氮氣保護下室溫避光反應0.5~48h,并提純;
(iii)將步驟(ii)提純后的化合物與0.001~10mmol馬來酸酐)加入0.3~300mL冰醋酸回流2~72h后2~8h內將0.001~10mmol乙酸酐逐滴地加入到混合溶液中,繼續回流0.5~72h,反應后提純;
(iv)將步驟(iii)所得化合物溶于0.2~200mL四氫呋喃中,并在4~37℃通氮氣攪拌的條件下與0.001~10mmol N-甲基嗎啡啉混合,接著逐滴地加入溶解于0.1~10mL四氫呋喃的0.001~10mmol氯甲酸異丁酯;5~60min后,逐滴地加入溶解于0.1~100mL四氫呋喃的0.001-10mmol叔丁基咔唑鹽,在4~37℃反應5~120min,接著在室溫反應0.5~48h;通過蒸發干燥的方法去除溶劑,并加入乙酸乙酯和水并使其分層,接著加入稀鹽酸,水和稀碳酸氫鈉對有機層,進行進一步的清洗并,通過無水硫酸鈉干燥,使溶劑蒸發;
(v)將步驟(iv)所得產物溶于0.1~100mL冰三氟醋酸中,并冰浴攪拌1~60min,在室溫高真空的環境下去除酸,加入乙醚后攪拌分散生成結晶產物,并提純;
(vi)將步驟(v)所得產物與0.001~10mmol含酮羰基或羧基的抗腫瘤藥物(例如含酮羰基的阿霉素)溶于0.1~100mL甲醇中,然后加入0.1~10mL三氟醋酸催化該反應,并在室溫避光攪拌0.5~48h;將反應后的產物在0~60℃減壓的條件下濃縮到0.1~100mL,然后加入0.1~100mL乙腈并于0~60℃放置0.5~48h使產物結晶,并離心收集沉淀,采用體積比為0~1:0~20的甲醇和乙腈溶液清洗沉淀并真空干燥得到本發明所述可注射的腫瘤靶向性熱敏前藥。
當步驟(i)中使用4,4'-偶氮雙(4-氰基戊酸,步驟(ii)中使用乙二胺時,步驟(ii)所得產物為HOOCCH2CH2C(CH3)(CN)N=NC(CH3)(CN)CH2CH2CONHCH2CH2NH2,步驟(iii)所得產物為C2H2(CO)2NCH2CH2NHCOCH2CH2C(CH3)(CN)N=NC(CH3)(CN)CH2CH2COOH,步驟(iv)所得產物為C2H2(CO)2NCH2CH2NHCOCH2CH2C(CH3)(CN)N=NC(CH3)(CN)CH2CH2CONHNH2,步驟(v)所得產物為C2H2(CO)2NCH2CH2NHCOCH2CH2C(CH3)(CN)N=NC(CH3)(CN)CH2CH2CONHNH2.CF3COOH,當步驟(vi)以阿霉素為原料時,所得到的本發明所述可注射的腫瘤靶向性熱敏前藥如下所示:
再一方面,本發明提供一種藥物組合物,該藥物組合物含治療有效量的本發明所述可注射的腫瘤靶向性熱敏前藥。
再一方面,本發明提供所述可注射的腫瘤靶向性熱敏前藥或所述藥物組合物在制備用于治療腫瘤的熱化療藥物中的應用。
本發明所述可注射的熱敏前藥或含其的組合物應用于熱化療,在具體應用時,當將治療有效量的熱敏前藥或藥物組合物給予受體后,需輔助熱療以達到治療目的,例如使用高頻電磁波、超聲波、熱水浴等使組織加熱,達到殺死癌細胞的溫度,致使腫瘤組織變形壞死,繼而使瘤體縮小或消除,在不損傷人體正常組織的情況下,以治療惡性腫瘤。
根據本發明的具體實施方式,本發明所述腫瘤包括結腸癌、直腸癌、腦瘤、肺癌、表皮鱗癌、膀胱癌、胰腺癌、乳腺癌、卵巢癌、宮頸癌、子宮內膜癌、結直腸癌、腎細胞癌、食管腺癌、食管鱗狀細胞癌、非霍奇金淋巴瘤、肝癌、皮膚癌、甲狀腺癌、頭頸癌、前列腺癌、神經膠質瘤及鼻咽癌中的一種或多種。
綜上所述,本發明主要提供了一種可注射的腫瘤靶向性熱敏前藥,該前藥主要應用于熱化療領域,該類前藥為熱響應前藥,其可通過熱響應機制可使馬來酸酐-藥物鍵合物在腫瘤部位定點釋放,大大增加藥物作用部位如細胞質和細胞核中的濃度,克服腫瘤的耐藥性,減少化療的毒副作用。本發明有助于推動熱化學治療的研究與發展,同時為癌癥的臨床診斷和治療提供新理論和新方法。
附圖說明
圖1為實施例6所得到的細胞毒性圖;
圖2為實施例7所得到的細胞毒性圖;
圖3為實施例7所得腫瘤生長曲線圖;
圖4為馬來酸酐-阿霉素鍵合物與阿霉素本身在水溶液中的溶解性測試實驗結果圖。
具體實施方式
為了對本發明的技術特征、目的和有益效果有更加清楚的理解,現結合具體實施例對本發明的技術方案進行以下詳細說明,應理解這些實例僅用于說明本發明而不用于限制本發明的范圍。實施例中,各原始試劑材料均可商購獲得,未注明具體條件的實驗方法為所屬領域熟知的常規方法和常規條件,或按照儀器制造商所建議的條件。
實施例1
(1)準確稱取0.00028g(0.001mmol)4,4'-偶氮雙(4-氰基戊酸),將其溶于0.1mL二甲基亞砜,再加入0.00012g(0.001mmol)NHS和0.00019g(0.001mmol)EDC及1μL吡啶,氮氣保護下室溫反應0.5h。
(2)將0.0000601g(0.001mmol)乙二胺加入上述反應溶液中,氮氣保護下室溫避光反應0.5h。
(3)采用過硅膠柱色譜分離的方法對產物進行提純并真空干燥。
(4)在步驟(3)干燥后的產物與0.000098g(0.001mmol)馬來酸酐中加入0.3mL冰醋酸回流2h,0.000102g(0.001mmol)乙酸酐逐滴地加入到這個混合溶液中2h,繼續回流0.5h。在10℃真空的環境下去除乙酸酐并形成一種固化的糖漿。并采用過硅膠柱色譜分離的方法進行提純。
(5)將步驟(4)所得產物溶于0.2mL四氫呋喃中,并在4℃通氮氣攪拌的條件下與0.000101g(0.001mmol)N-甲基嗎啡啉混合,接著逐滴地加入溶解于0.1mL四氫呋喃的0.000136g(0.001mmol)氯甲酸異丁酯,5min后,逐滴地加入溶解于0.1mL四氫呋喃的0.000132g(0.001mmol)叔丁基咔唑鹽,在4℃反應5min,接著在室溫反應0.5h。通過蒸發干燥的方法去除溶劑,并加入乙酸乙酯和水并使其分層。接著加入稀鹽酸、水和稀碳酸氫鈉對有機層進行進一步的清洗并通過無水硫酸鈉方法干燥,使溶劑蒸發得該步產物。
(6)將步驟(4)所得產物溶于0.1mL冰三氟醋酸中,并冰浴攪拌1min。在室溫高真空的環境下去除酸,加入乙醚后攪拌分散生成結晶產物。采用過硅膠柱色譜分離的方法進行提純產物。
(7)將步驟(6)所得產物和0.00054g(0.001mmol)鹽酸阿霉素溶于0.1mL甲醇中,然后加入0.1mL三氟醋酸去催化,在室溫避光攪拌0.5h。該混合物在0~60℃減壓的條件下濃縮到0.1mL。然后加入0.1mL乙腈并0℃放置0.5~48h使產物結晶,并離心收集沉淀。采用體積比為1:10的甲醇和乙腈混合溶液清洗沉淀并真空干燥得到產物。
實施例2
(1)準確稱取0.028g(0.1mmol)4,4'-偶氮雙(4-氰基戊酸),將其溶于10mL二甲基亞砜,再加入0.012g(0.1mmol)NHS和0.038g(0.2mmol)EDC及100μL吡啶,氮氣保護下室溫反應24h。
(2)將0.00601g(0.1mmol)乙二胺加入上述反應溶液中,氮氣保護下室溫避光反應0.5h。
(3)采用過硅膠柱色譜分離的方法對產物進行提純并真空干燥。
(4)在步驟(3)干燥后的產物與0.0098g(0.1mmol)馬來酸酐中加入30mL冰醋酸回流8h,0.0102g(0.1mmol)乙酸酐逐滴地加入到這個混合溶液中1h,繼續回流4h。在40℃真空的環境下去除乙酸酐并形成一種固化的糖漿。并采用過硅膠柱色譜分離的方法進行提純。
(5)將步驟(4)所得產物溶于20mL四氫呋喃中,并在4℃通氮氣攪拌的條件下與0.0101g(0.1mmol)N-甲基嗎啡啉混合,接著逐滴地加入溶解于10mL四氫呋喃的0.0136g(0.1mmol)氯甲酸異丁酯,30min后,逐滴地加入溶解于10mL四氫呋喃的0.0132g(0.1mmol)叔丁基咔唑鹽,在4℃反應5min,接著在室溫反應2h。通過蒸發干燥的方法去除溶劑,并加入乙酸乙酯和水并使其分層。接著加入稀鹽酸、水和稀碳酸氫鈉對有機層進行進一步的清洗并通過無水硫酸鈉方法干燥,使溶劑蒸發得該步產物。
(6)將步驟(4)所得產物溶于10mL冰三氟醋酸中,并冰浴攪拌5min。在室溫高真空的環境下去除酸,加入乙醚后攪拌分散生成結晶產物。采用過硅膠柱色譜分離的方法進行提純產物。
(7)將步驟(6)所得產物和0.054g(0.1mmol)鹽酸阿霉素溶于10mL甲醇中,然后加入10mL三氟醋酸去催化,在室溫避光攪拌10h。該混合物在30℃減壓的條件下濃縮到10mL。然后加入10mL乙腈并30℃放置0.5~48h使產物結晶,并離心收集沉淀。采用體積比為1:10的甲醇和乙腈混合溶液清洗沉淀并真空干燥得到產物。
實施例3
(1)準確稱取2.8g(10mmol)4,4'-偶氮雙(4-氰基戊酸),將其溶于100mL二甲基亞砜,再加入1.2g(10mmol)NHS和1.9g(10mmol)EDC及1000μL吡啶,氮氣保護下室溫反應48h。
(2)將0.601g(10mmol)乙二胺加入上述反應溶液中,氮氣保護下室溫避光反應48h。
(3)采用過硅膠柱色譜分離的方法對產物進行提純并真空干燥。
(4)在步驟(3)干燥后的產物與0.98g(10mmol)馬來酸酐中加入300mL冰醋酸回流72h,1.02g(10mmol)乙酸酐逐滴地加入到這個混合溶液中8h,繼續回流72h。在80℃真空的環境下去除乙酸酐并形成一種固化的糖漿。并采用過硅膠柱色譜分離的方法進行提純。
(5)將步驟(4)所得產物溶于200mL四氫呋喃中,并在4℃通氮氣攪拌的條件下與1.01g(10mmol)N-甲基嗎啡啉混合,接著逐滴地加入溶解于10mL四氫呋喃的1.36g(10mmol)氯甲酸異丁酯,5min后,逐滴地加入溶解于100mL四氫呋喃的1.32g(10mmol)叔丁基咔唑鹽,在4℃反應120min,接著在室溫反應48h。通過蒸發干燥的方法去除溶劑,并加入乙酸乙酯和水并使其分層。接著加入稀鹽酸、水和稀碳酸氫鈉對有機層進行進一步的清洗并通過無水硫酸鈉方法干燥,使溶劑蒸發得該步產物。
(6)將步驟(4)所得產物溶于100mL冰三氟醋酸中,并冰浴攪拌60min。在室溫高真空的環境下去除酸,加入乙醚后攪拌分散生成結晶產物。采用過硅膠柱色譜分離的方法進行提純產物。
(7)將步驟(6)所得產物和5.4g(10mmol)鹽酸阿霉素溶于100mL甲醇中,然后加入10mL三氟醋酸去催化,在室溫避光攪拌48h。該混合物在60℃減壓的條件下濃縮到100mL。然后加入100mL乙腈并60℃放置48h使產物結晶,并離心收集沉淀。采用體積比為1:10的甲醇和乙腈混合溶液清洗沉淀并真空干燥得到產物。
實施例4
(1)準確稱取0.028g(0.1mmol)4,4'-偶氮雙(4-氰基戊酸),將其溶于10mL二甲基亞砜,再加入0.012g(0.1mmol)NHS和0.038g(0.2mmol)EDC及100μL吡啶,氮氣保護下室溫反應24h。
(2)將0.00601g(0.1mmol)乙二胺加入上述反應溶液中,氮氣保護下室溫避光反應0.5h。
(3)采用過硅膠柱色譜分離的方法對產物進行提純并真空干燥。
(4)在步驟(3)干燥后的產物與0.0098g(0.1mmol)馬來酸酐中加入30mL冰醋酸回流8h,0.0102g(0.1mmol)乙酸酐逐滴地加入到這個混合溶液中1h,繼續回流4h。在40℃真空的環境下去除乙酸酐并形成一種固化的糖漿。并采用過硅膠柱色譜分離的方法進行提純。
(5)將步驟(4)所得產物溶于20mL四氫呋喃中,并在4℃通氮氣攪拌的條件下與0.0101g(0.1mmol)N-甲基嗎啡啉混合,接著逐滴地加入溶解于10mL四氫呋喃的0.0136g(0.1mmol)氯甲酸異丁酯,30min后,逐滴地加入溶解于10mL四氫呋喃的0.0132g(0.1mmol)叔丁基咔唑鹽,在4℃反應5min,接著在室溫反應2h。通過蒸發干燥的方法去除溶劑,并加入乙酸乙酯和水并使其分層。接著加入稀鹽酸、水和稀碳酸氫鈉對有機層進行進一步的清洗并通過無水硫酸鈉方法干燥,使溶劑蒸發得該步產物。
(6)將步驟(4)所得產物溶于10mL冰三氟醋酸中,并冰浴攪拌5min。在室溫高真空的環境下去除酸,加入乙醚后攪拌分散生成結晶產物。采用過硅膠柱色譜分離的方法進行提純產物。
(7)將步驟(6)所得產物和0.0564g(0.1mmol)柔紅霉素溶于10mL甲醇中,然后加入10mL三氟醋酸去催化,在室溫避光攪拌10h,所得混合物在30℃減壓的條件下濃縮到10mL。然后加入10mL乙腈并30℃放置0.5~48h使產物結晶,并離心收集沉淀。采用體積比為1:10的甲醇和乙腈混合溶液清洗沉淀并真空干燥得到產物。
實施例5
(1)準確稱取0.028g(0.1mmol)4,4'-偶氮雙(4-氰基戊酸),將其溶于10mL二甲基亞砜,再加入0.012g(0.1mmol)NHS和0.038g(0.2mmol)EDC及100μL吡啶,氮氣保護下室溫反應24h。
(2)將0.00601g(0.1mmol)乙二胺加入上述反應溶液中,氮氣保護下室溫避光反應0.5h。
(3)采用過硅膠柱色譜分離的方法對產物進行提純并真空干燥。
(4)在步驟(3)干燥后的產物與0.0098g(0.1mmol)馬來酸酐中加入30mL冰醋酸回流8h,0.0102g(0.1mmol)乙酸酐逐滴地加入到這個混合溶液中1h,繼續回流4h。在40℃真空的環境下去除乙酸酐并形成一種固化的糖漿。并采用過硅膠柱色譜分離的方法進行提純。
(5)將步驟(4)所得產物溶于20mL四氫呋喃中,并在4℃通氮氣攪拌的條件下與0.0101g(0.1mmol)N-甲基嗎啡啉混合,接著逐滴地加入溶解于10mL四氫呋喃的0.0136g(0.1mmol)氯甲酸異丁酯,30min后,逐滴地加入溶解于10mL四氫呋喃的0.0132g(0.1mmol)叔丁基咔唑鹽,在4℃反應5min,接著在室溫反應2h。通過蒸發干燥的方法去除溶劑,并加入乙酸乙酯和水并使其分層。接著加入稀鹽酸、水和稀碳酸氫鈉對有機層進行進一步的清洗并通過無水硫酸鈉方法干燥,使溶劑蒸發得該步產物。
(6)將步驟(4)所得產物溶于10mL冰三氟醋酸中,并冰浴攪拌5min。在室溫高真空的環境下去除酸,加入乙醚后攪拌分散生成結晶產物。采用過硅膠柱色譜分離的方法進行提純產物。
(7)將步驟(6)所得產物和0.0454g g(0.1mmol)甲氨蝶呤溶于10mL甲醇中,然后加入10mL三氟醋酸去催化,在室溫避光攪拌24h,所得混合物在30℃減壓的條件下濃縮到10mL。然后加入10mL乙腈并30℃放置24h使產物結晶,并離心收集沉淀。采用體積比為1:10的甲醇和乙腈混合溶液清洗沉淀并真空干燥得到產物。
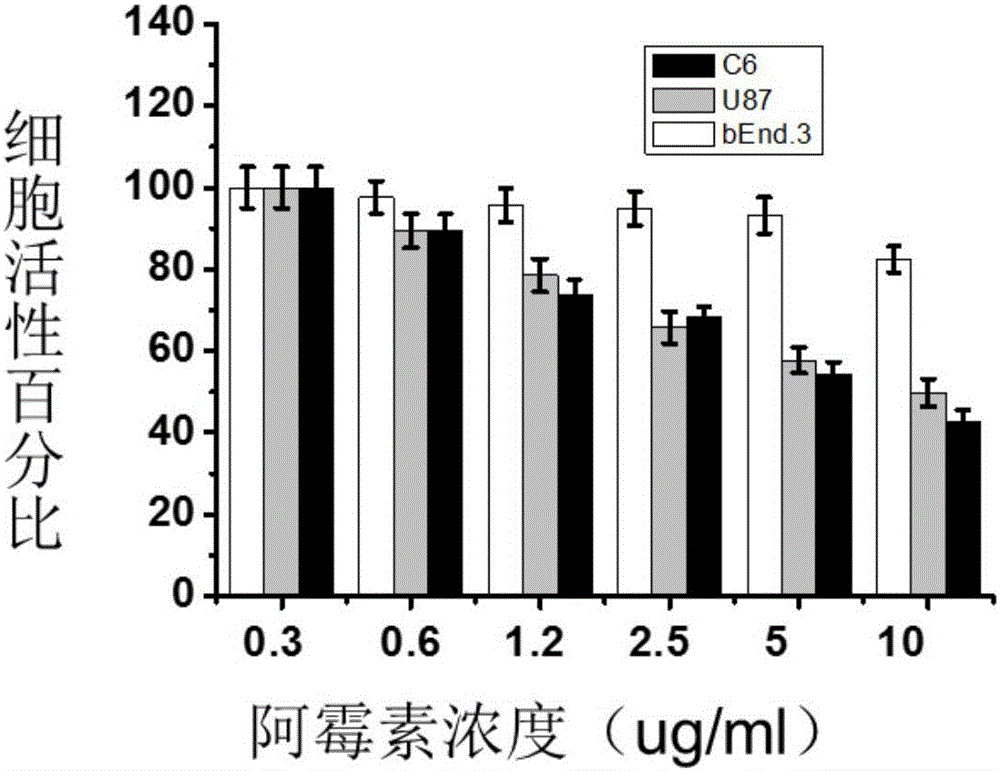
實施例6:比較馬來酸酐-阿霉素鍵合物對兩株腦膠質瘤細胞C6,U87和內皮細胞bEnd.3的細胞毒性作用。
本實施例使用的C6,U87,bEnd.3皆屬于腦膠質瘤研究中常用細胞株,均能通過市場購買獲得。將實施例2獲得的馬來酸酐-阿霉素鍵合物用DMEM培養基等比例稀釋成適當濃度,采用噻唑藍(MTT)快速比色法測定馬來酸酐-阿霉素鍵合物對C6,U87,bEnd.3的細胞毒性作用。
將對數生長期的C6,U87,bEnd.3細胞以1~5×104個/孔加入96孔板中,過夜培養至貼壁,然后分別用含有相應濃度的馬來酸酐-阿霉素鍵合物DMEM培養基在37℃培養48h,每孔加入20μl MTT(5mg/ml),37℃,5%CO2及飽和濕度的培養箱中孵育4h,吸出培養基,加入150μl DMSO,10min后490nm酶標儀檢測,并繪制細胞毒性圖,所得結果如圖1所示,從圖1中可以看到明顯對細胞的無明顯殺傷作用,證明馬來酸酐-阿霉素鍵合物在正常溫度下對細胞無毒。
實施例7:馬來酸酐-阿霉素鍵合物與熱療聯合作用。
本實施例使用的C6,U87,bEnd.3皆屬于腦膠質瘤研究中常用細胞株,均能通過市場購買獲得。將實施例2獲得的馬來酸酐-阿霉素鍵合物用DMEM培養基等比例稀釋成適當濃度,采用噻唑藍(MTT)快速比色法測定阿霉素對C6,U87,bEnd.3的細胞毒性作用。
將對數生長期的C6,U87,bEnd.3細胞以1~5×104個/孔加入96孔板中,過夜培養至貼壁,然后分別用含有相應濃度的馬來酸酐-阿霉素鍵合物DMEM培養基于37℃培養47h 42℃培養1h,每孔加入20μl MTT(5mg/ml),37℃,5%CO2及飽和濕度的培養箱中孵育4h,吸出培養基,加入150μl的DMSO,10min后490nm酶標儀檢測,并繪制細胞毒性圖,所得結果如圖2所示,對比圖1及圖2可以看出馬來酸酐-阿霉素鍵合物對C6,U87細胞的有明顯殺傷作用,對bEnd.3細胞的無明顯殺傷作用,證明白蛋白-阿霉素鍵合物在中溫條件下對正常無毒,而對腫瘤細胞存在明顯的毒性。
實施例8
本實施例動物腫瘤模型熱化療聯合治療實驗的具體方案:24只6~8周裸鼠(Balb/c),6只一組,共分4個實驗組腦膠質瘤進行實體腫瘤的建模,4個實驗組分別為:生理鹽水組(陰性對照);聚焦超聲組(溫度控制在42℃,20min);馬來酸酐-阿霉素鍵合物組;馬來酸酐-阿霉素鍵合物和聚焦超聲組(溫度控制在42℃,20min)。在腫瘤模型長至50mm3后,將實施例2獲得的馬來酸酐-阿霉素鍵合物溶液按每Kg老鼠2mg的用量通過尾靜脈注射到小鼠體內,采用聚焦超聲選擇性對小鼠腫瘤組織進行超聲,觀察腫瘤生長狀況并繪制腫瘤生長曲線,所得結果如圖3所示,從圖3中可以看出馬來酸酐-阿霉素鍵合物和聚焦超聲組較其他組可顯著抑制腫瘤生長。
配制0.1mg/ml(以阿霉素的量計算)的阿霉素和馬來酸酐-阿霉素鍵合物(實施例3)的PBS7.4的水溶液,于24小時候用照相機對這兩種溶液進行拍照,所得結果如圖4所述,從圖4中可以看出阿霉素發生了沉淀,而馬來酸酐-阿霉素鍵合物在水中的呈透明狀,溶解性顯著提高。
最后說明的是:以上實施例僅用于說明本發明的實施過程和特點,而非限制本發明的技術方案,盡管參照上述實施例對本發明進行了詳細說明,本領域的普通技術人員應當理解:依然可以對本發明進行修改或者等同替換,而不脫離本發明的精神和范圍的任何修改或局部替換,均應涵蓋在本發明的保護范圍當中。
- 還沒有人留言評論。精彩留言會獲得點贊!