一種紫杉醇納米粒及其制備方法與流程
本發明涉及一種高分子納米顆粒的制備方法,尤其涉及一種紫杉醇納米粒及其制備方法。
背景技術:
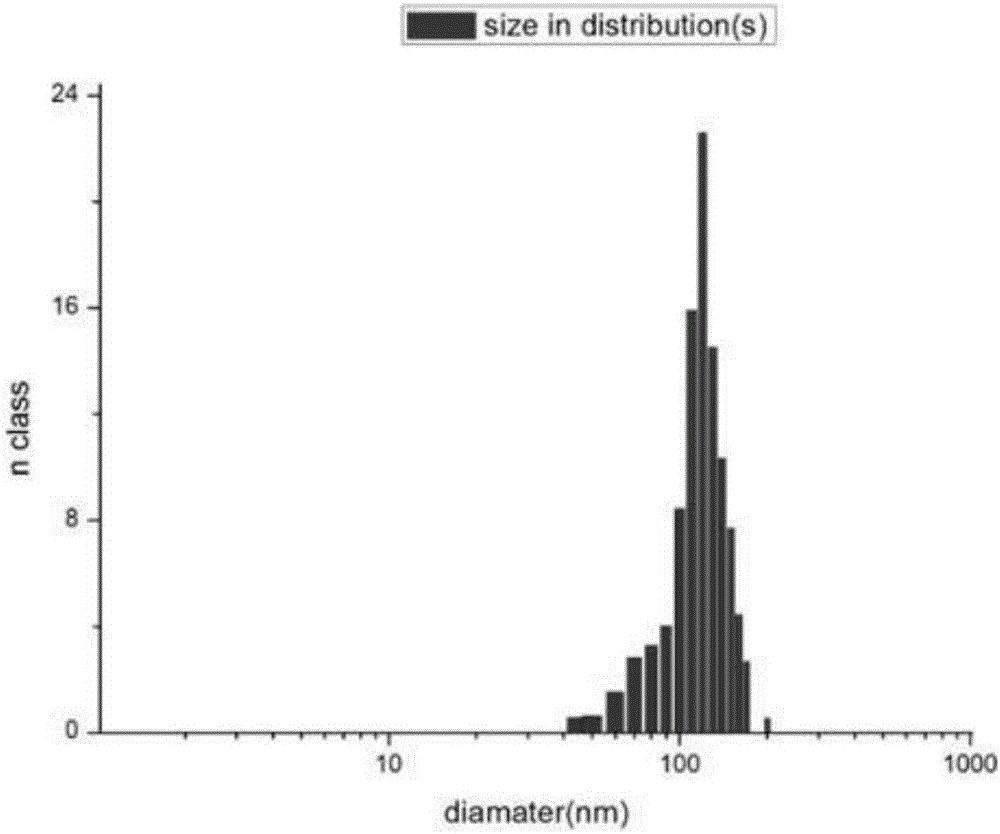
:紫杉醇(Paclitaxel)是從太平洋紫杉(Taxusbrevifolia)樹皮中提取分離的二萜類化合物,難溶于水,紫杉醇抗腫瘤機制主要是通過抑制細胞微管蛋白活性從而具有細胞毒性,是卵巢癌和乳腺癌治療的一線用藥,然而多藥耐藥,溶解性差以及化療毒性限制了其臨床療效。紫杉醇臨床使用的注射劑大多是將紫杉醇增溶于聚氧乙烯蓖麻油(CremophorEL)無水乙醇(1∶1)中,臨用前稀釋至給藥濃度。但聚氧乙烯蓖麻油會引起嚴重的過敏反應,患者給藥前要預先服用抗過敏藥物以減輕不良反應。限制了其在臨床上的應用。為改善紫杉醇的溶解性,目前主要有兩種方式,一是化學修飾,二是物理包裹;其中,化學修飾還不能夠使紫杉醇在治療效果不降低,毒性不增強的情況下,增加其自身的溶解性,提高抗腫瘤的效率。納米科技從在20世紀90年代初興起到現在經歷了飛速的發展,取得了令人矚目的成果。目前納米技術已經廣泛的應用于航空航天、生物醫學、材料等多個領域。在醫藥領域方面,載藥納米載體的出現和發展,為靶向抗腫瘤藥物的研制提供了新的途徑。應用納米載體包裹藥物,可以增加難溶性藥物的溶解度,加強水溶性藥物的穩定性,提高藥物的生物利用度,減少藥物的毒副作用。紫杉醇被包裹入納米載體系統中,其在體內的生物學分布將發生改變,其中與載藥納米載體的生物學分布密切相關。納米載體通過循環系統進入血液后,大部分與血液中的蛋白調理素相結合,被網狀內皮系統(RSE)所識別,最先被肝臟脾臟等器官所攝取,其在血液中的濃度下降,僅有少部分可以經過血液循環系統到達腫瘤組織中。研究表明由于腫瘤組織生長快,血供充足,其腫瘤組織中血管的通透性較正常血管高,同時腫瘤組織缺乏有效的淋巴引流,因此載藥納米載體對腫瘤組織具有增強的滲透性和滯留性效應(EPR效應),從而達到對腫瘤組織的被動靶向及緩釋作用。HPH敏感性控釋(物理化學靶向)腫瘤組織無氧酵解增加,腫瘤局部呈現酸性環境,使用PH敏感性的生物可降解材料制備的納米載體,使納米載體選擇性的在腫瘤部位降解釋放藥物,而在正常組織中保持穩定,可實現腫瘤的靶向。聚乳酸可在腫瘤組織酸性環境中降解,釋放藥物,從而實現了納米載體的物理化學靶向性。納米載體在體內的代謝與納米載體本身的性質入粒徑、電荷、表面的化學修飾以及納米的結構和組成等方面的因素有關。理想的納米載體粒徑應該保持在100nm左右,電位在10mV以內,并且在表面進行一定的化學修飾,進一步篩選腫瘤特異性或高表達的受體,抗原,將相應的配體或者是特異性的單克隆抗體與納米載體通過物理或者化學鍵的方式相耦聯,依靠靶向分子特異性識別腫瘤組織,從而提高對腫瘤組織的選擇性,減少載藥納米載體在周圍組織中的聚集,將實現對藥物的主動靶向傳遞作用,使紫杉醇納米載體在腫瘤組織中的蓄積。目前常見的靶向分子主要是特異性或者是腫瘤組織高表達的單克隆抗體以及受體配基。但是大多是抗體局限在細胞膜上,無法實現藥物的細胞內傳遞,同時容易引起體內的免疫排除反應,加上抗體價格高昂,使得抗體的應用受到了很大的限制。受體配基修飾的納米載體,由于配體與受體之間特異性強,親和力高,并且不易引起免疫反應等優點,引起了人們的廣泛關注,其中最具有代表性的是葉酸。葉酸(FA)是一種水溶性B族維生素,是人體必須的一種營養物質,參與人體多項重要物質的合成和代謝。在真核細胞中參與核苷酸的生物合成,是細胞生存和增殖所必須的營養物質。腫瘤細胞中葉酸受體大量表達,如卵巢癌、乳腺癌、結腸癌、肺癌、前列腺癌及鼻喉和腦部的腫瘤等,顯著高于正常組織細胞葉酸受體表達水平。葉酸對腫瘤組織有特異性和靶向性,且本身為小分子物質,水溶性好,能在各種溶液中穩定存在,易儲存,且對人體沒有毒副作用,是一個理想的靶向分子。納米載體可以改變疏水性紫杉醇的水溶性,更好的符合靜脈給藥的方式,提高紫杉醇的生物利用度。此外,葉酸靶向納米載體可以通過主動靶向,被動靶向,以及PEG-PLA納米載體的物理靶向性作用提高腫瘤組織的富集能力,增加腫瘤中紫杉醇的含量。然而納米載體自身的緩釋作用也會維持腫瘤組織中的紫杉醇含量在有效的范圍內。因此,具有靶向性的納米載體與傳統的化療藥紫杉醇聯合,可以更好的體現腫瘤的靶向治療,提高治療的效果。目前納米紫杉醇顆粒的制備方法普遍存在以下問題:為了得到紫杉醇高分子納米顆粒的懸浮液,需要反復多次的高速離心-重懸,步驟繁瑣;在制備水相的過程中,需要加入表面活性劑,表面活性劑的加入會導致納米顆粒在使用的過程中增加對胃腸道的刺激性;藥物制備成功后不經過凍存除去有機溶劑,直接注入體內,有機溶劑對機體造成傷害,并且不易儲存;凍存后的納米粒復溶存在溶解性差、形態和粒徑大小會發生改變等缺點。技術實現要素:本發明的目的之一是提供一種紫杉醇納米粒的制備方法。本發明的目的之二是提供一種紫杉醇納米粒。本發明的目的之三是提供一種治療腫瘤的藥物的制備方法,包括本發明中所述的紫杉醇納米粒的制備方法,優選:所述的腫瘤為卵巢癌、乳腺癌、結腸癌、肺癌、前列腺癌、鼻喉或腦部的腫瘤。為了實現上述目的,本發明采用如下技術方案:一種紫杉醇納米粒的制備方法,步驟如下:(1)將納米嵌段聚合物聚乙二醇-聚乳酸(PEG-PLA)或者是聚乙二醇-聚乳酸(PEG-PLA)和葉酸-聚乙二醇-聚乳酸(folate-PEG-PLA)的混合聚合物溶解在有機溶劑四氫呋喃中,制成溶液A;(2)將紫杉醇溶于二氯甲烷中,制成溶液B;(3)將溶液A和溶液B充分混合,制成濃度均一的藥物聚合物有機溶液;(4)將步驟(3)制備的混合溶液中加入質量分數為2%的聚乙烯醇,超聲使混合溶液充分混合;(5)以20-30滴/分鐘的速度逐滴將混合后的溶液滴入水中,冰浴下超聲;(6)將所得的乳液常溫常壓下水浴攪拌,揮發有機溶劑,離心,收集沉淀,用水洗滌,重新水化;(7)將重新水化后的膠束溶液放入透析袋中,將透析袋置于水中進行透析,即得到紫杉醇納米膠束;(8)向透析好的納米膠束加入質量分數為2.5%的凍干保護劑為F-68,凍存得到紫杉醇納米粒;所述納米嵌段聚合物與紫杉醇的質量比為:6:1。優選的:所述步驟(1)中當納米嵌段聚合物為PEG-PLA和folate-PEG-PLA的混合聚合物時,folate-PEG-PLA的質量百分比為8%-12%,更優選為10%。優選的:所述步驟(1)中當納米嵌段聚合物為聚乙二醇-聚乳酸的分子量為10000-13000,聚合物中乙二醇:乳酸單體的摩爾比例為(4-5):1。優選的:所述步驟(4)中,聚乙烯醇與混合溶液的體積比為1.5:1;超聲的功率為30-40W,超聲的時間為10min。優點:該條件下兩種溶液能夠充分的混合,并且要求所制備的納米膠束分散程度好,顆粒呈均勻的球形。優選的:所述步驟(6)中,攪拌采用磁力攪拌器攪拌,攪拌速度為500-1000r/min,攪拌時間為4h。優點:攪拌可以改變溶液中各成分濃度的分布,有利于各種成分的分散,攪拌速度較低時溶液不能充分的反應,納米粒成形不好,容易產生厚膜,攪拌速度過高,納米粒容易遭到破壞,所以適宜的攪拌速度可以使溶液中的成分相對均勻,形成的納米粒大小均勻,粒徑合適。所以本發明選用最適宜的攪拌速度為500-1000rpm/min。攪拌時間對納米粒的形態有關,攪拌時間過短,有機溶劑揮發不完全,納米粒形態不規則,分子量較少。攪拌時間過長,納米粒容易遭到破壞,不利于納米粒工藝的生產。因此,最佳的攪拌時間為4h。優選的:所述步驟(6)中,離心的條件為14000r/min離心時間為30min。優點:通過離心可以有效的收集納米粒,離心轉速太慢,離心時間太短則收集不完全,離心過快,離心時間長,則納米粒形態遭到破壞,不能實現有效的載藥,所以通過篩選,本發明的離心時間為30min,轉速為14000r/min,既保證完全收集了納米粒,又避免了納米粒形態的破壞。優選的:所述步驟(7)中,透析袋的截留分子量為3500KDa。本發明的有益效果:1、本發明將制備的紫杉醇納米膠束加入保護劑凍干,凍干后呈疏松的粉末狀,復溶后所得的納米粒性質沒有發生改變,粒徑為100-150nm,納米粒呈球形分散佳,紫杉醇包裹在納米粒之中,減少了其他組織對于紫杉醇的攝取,從而減少了紫杉醇帶來的毒副作用,復溶后檢測藥物的體外釋放情況和凍干前一樣,說明本發明方法制備的紫杉醇納米粒性質穩定,將藥物膠束溶液在常溫下放置一周,無藥物釋放,進一步證實藥物包裹在納米顆粒的核心,并且性質較為穩定。2、本發明的方法所制備的紫杉醇納米粒載藥量為40%,包封率為83%,載藥量和包封率均較高,減少了紫杉醇的用量,具有極高的應用價值。3、當納米嵌段聚合物為PEG-PLA和folate-PEG-PLA的混合聚合物時,將不同葉酸含量的納米粒與卵巢癌細胞SKOV3孵育后,用熒光顯微鏡觀察,當folate-PEG-PLA質量百分比小于8%時,隨著葉酸含量的增加,細胞內熒光的強度也隨之增加,當混合物中folate-PEG-PLA質量百分比大于12%時,隨著葉酸含量的增加,熒光強度不會增加,由此說明,混合物中葉酸質量百分數在8-12%時可以達到最佳的治療效果。4、聚乙二醇-聚乳酸為兩親性聚合物,其組成的納米粒既可以有效的溶于水溶液,其內部疏水區域可以為脂溶性藥物提供空間,從而提高紫杉醇的水溶性,改變其在體內的分布,從而提高藥物的療效。聚乳酸可以在體內分解為單體乳酸,并且在肝臟中轉化為葡萄糖提供能量。而聚乙二醇是美國食品藥品管理局(FDA)批準可用于人體的高分子化合物。葉酸是一種人體重要的營養物質,參與人體多種代謝反應。因此,上述材料制備的載藥納米粒對人體毒副作用較小。此外,本發明采用的聚乙二醇-聚乳酸聚合物有端氨基,可以通過化學鍵鏈接葉酸基團,增加了納米了的腫瘤靶向性作用。聚乙二醇-聚乳酸的分子量為10000-13000,聚合物中乙二醇:乳酸單體的摩爾比例為(4-5):1,聚合物的分子量,以及聚合物中輸水部分和親水部分的質量的比例關系,會直接影響到聚合物的膠束形態,本發明通過對聚合物分子量以及對乙二醇和乳酸比例的優化,制備出的納米粒形態均一呈球形,分散好。5、本發明通過對多種不同的有機溶解進行考察,結果發現,應用四氫呋喃溶解納米嵌段聚合物,所制備的納米膠束形態規則,分散狀態佳,所得納米粒的粒徑在120mn左右,較其他有機溶劑獲得的膠束溶液狀態好。用二氯甲烷溶解紫杉醇,可以提高紫杉醇的溶解性,增加載藥納米載體的包封率和載藥量。步驟(4)中,有機溶劑為質量分數為2%聚乙烯醇,可以使混合溶液充分的混合。6、藥物聚合物有機溶劑的滴加速度對膠束的形成產生重要的影響,滴加的速度過快,反應不完全,形成的膠束粒徑大,粒度分布廣,并且不穩定,常有一層厚膜。滴加速度過慢,會使納米粒數量較少,生產周期延長。掌握好藥物集合物有機溶液的滴加速度對保證制備膠束的工藝穩定性和產品性能有重要的影響。因此本發明通過不斷的優化,篩選出最佳的滴速為20-30滴/分鐘。7、納米粒凍干保護劑主要的作用為保護納米粒結構的完整,防止納米粒在凍干保存的過程中團聚,增加凍干后納米粒的溶解性。通過不斷的篩選發現,不加保護劑或者是加入其他類型的保護劑如甘露醇等,凍干后的納米粒溶解性,形態和粒徑大小均發生改變。因此,本發明選用的凍干保護劑為質量分數為2.5%的F-68,避免了上述問題的出現。8、紫杉醇和聚合物不易溶于水,分別用有機溶劑PEG-PLA或者是PEG-PLA和folate-PEG-PLA的混合聚合物與二氯甲烷分別進行溶解形成油相,再加入不溶于有機溶劑的聚乙烯醇(易溶于水)形成水相,利用超聲形成均一的乳液,有利于納米粒的形成。附圖說明圖1為紫杉醇納米膠束透射電鏡圖;圖2為紫杉醇納米膠束粒徑分布圖;圖3為紫杉醇納米粒凍干復溶后的透射電鏡圖;圖4為紫杉醇納米粒凍干復溶后的粒徑分布圖。具體實施方式下面結合附圖與實施例對本發明作進一步說明。實施例1:紫杉醇納米粒制備條件的優化1.制備方法的考察納米粒的制備可以采取旋轉蒸發法、透析法和旋轉蒸發透析法。旋轉蒸發法是將藥物與納米材料混合后的溶液滴加到水中,以一定的速度攪拌獲得膠束溶液。而透析法則將藥物和納米嵌段直接裝到透析袋中,透過透析獲得納米膠束溶液。旋轉蒸發透析法則是將兩者結合,旋轉蒸發的方法同前述,將旋轉蒸發所得的溶液裝入透析袋中,在水中透析,從而獲得膠束溶液。旋轉蒸發法的優勢在于反應較為徹底,獲得較為純凈的納米粒。通過透射電鏡對不同方法制備的納米粒進行觀察,結果見表1.表1不同制備方法對納米粒形態的影響制備方法納米粒形態旋轉蒸發法粒徑大小不均勻,易形成厚膜透析法形態不規則,分散佳旋轉蒸發透析法形態規則呈球形,大小均勻,分散佳2.有機溶劑的考察分別精密稱取等量的PEG-PLA納米嵌段共4四份,分別用溶于等量的不同的有機溶液丙酮,四氫呋喃,DMSO以及二甲基甲酰胺中。準備4個50ml燒杯,分別加入等量的超純水,在相同的攪拌速度下,將含有納米材料的不同有機溶液以20-30滴/分鐘的速度逐滴滴加到超純水中,繼續攪拌,將攪拌好的膠束溶液放入透析袋中。透析相同的時間,取適量的假體通過透射電鏡觀察其形態,篩選出合適的有機溶劑。應用四氫呋喃有機溶劑制備的納米膠束形態均一,分散性好,粒徑在100nm左右,而其它有機溶劑獲得的膠束粒徑較小,或者不能形成納米粒,因此可以選擇四氫呋喃作為納米嵌段的有機溶劑,結果見表2表2不同有機溶劑對納米粒形態的影響有機溶劑納米粒形態丙酮粒徑大小為50nm左右,分散好,顆粒大小均勻四氫呋喃顆粒形態規則呈球形,粒徑在100nm左右,分散好DMSO不易形成納米顆粒二甲基甲酰胺成形不佳,粒徑在40nm左右,以粘連,形成厚膜3.聚乙烯醇的用量對納米形態的影響分別精密稱取PEG-PLA聚合物300mg,均溶于1ml四氫呋喃中,制備成濃度為300mg/ml的聚合物有機溶液,稱取紫杉醇50mg溶于1ml二氯甲烷中,將兩者混合后加入2ml,3ml,4ml,5ml聚乙烯醇,超聲充分,以20-30滴/分鐘的速度滴加到30ml純水中,繼續攪拌兩個小時,將攪拌后的溶液放入透析袋中,透射電鏡觀察納米粒的形態,當聚乙烯醇的量為2ml時,超聲后不能夠形成均一的溶液,有塊狀物沒有充分反應,制備的納米粒形成厚膜,聚乙烯醇用量太多,所得的納米粒量少,分散不佳。聚乙烯醇的用量對納米形態的影響見下表。聚乙烯醇用量納米形態2ml形成厚膜3ml納米呈球形,易分散4ml納米粒少呈球形5ml納米粒少呈球形4.藥物聚合物有機溶液/水的比例對納米形態的影響分別精密稱取PEG-PLA聚合物300mg,均溶于1ml四氫呋喃中,制備成濃度為300mg/ml的聚合物有機溶液,稱取紫杉醇50mg溶于1ml二氯甲烷中,加入3ml聚乙烯醇,超聲充分混合。分別在4個燒杯中加入10ml、20ml、30ml、40ml超純水,保證有機溶液/水比例為1:2、1:4、1:6、1:8,在一定速度磁力攪拌的情況下,將含有納米材料的不同濃度的有機溶液以20-30滴/分鐘的速度逐滴加入到純水中,繼續攪拌后放入透析袋中透析,獲得載藥納米膠束后取適量的納米膠束經透射電鏡觀察其形態。根據形態可見,不同有機溶液/水比例對納米粒形態的影響并不是很大,當有機溶劑/水比例<1:8時,制備納米膠束形態規則呈球形,分散佳。見表4表4藥物聚合物有機溶液/水的比例對納米粒形態的影響藥物聚合物有機溶液/水納米粒形態1:2形態規則,有厚膜1:4形態規則呈球形,粘連多1:6形態規則,顆粒多,分散佳1:8成形不佳,粒徑在40nm左右,顆粒較少5.攪拌速度對納米粒形態的影響分別稱取PEG-PLA共三份,溶于一定量的四氫呋喃中,在另一個50ml燒杯中加入一定量的超純水,在不同速度磁力攪拌速度的情況下,將含有納米材料的有機溶劑以20-30滴/分鐘的速度逐滴滴加到純水中,繼續攪拌一段時間后放入透析袋中,透析得到的納米膠束經過透射電鏡檢測觀察其形態。根據形態分析,不同的攪拌速度對納米粒形態的影響很大,選擇合適的攪拌速度制備的納米載體形態均一,分散性好。表5攪拌速度對納米粒的影響6.凍干保護劑對納米粒形態的影響將上述制備成功的納米膠束不加保護劑或者加入不同的凍干保護劑,觀察凍干后溶解性以及復溶后透射電鏡結果如表5所示。表6加入不同的保護劑對納米粒形態的影響不同凍干保護劑凍干后外觀納米粒形態不加保護劑顏色乳白色,不復溶不溶于水,溶于有機溶劑5%甘露醇顏色白色,水溶性好納米粒形態改變,較多的粘連2.5%F-68顏色乳白色,水溶性好納米粒形態未發生改變實施例2:無葉酸紫杉醇納米粒的制備精密稱取PEG-PLA300mg聚合物,溶于1ml四氫呋喃中,配制成300mg/ml的有機溶液。避光稱取紫杉醇50mg,溶于1ml的二氯甲烷中,配成濃度為50mg/ml的有機溶液。將兩者溶液充分混合制成均一的溶液,向該混合溶液中加入3ml聚乙烯醇,30-40W超聲10min,使藥物聚合物溶液充分的混合,將混合好的有機溶液以20-30滴/分鐘的速度滴加到30ml超純水中,冰浴300W超聲20min。將所得的乳液常溫常壓下水浴攪拌4h,揮發有機溶劑,離心(14000r/min)30min,收集沉淀,用蒸餾水洗滌4次,重新水化。將重新水化后的膠束溶液放入透析帶(3500KD)中將透析袋置于水中進行透析,將沒有完全反應的有機溶解和藥物透析出來,即得到紫杉醇納米膠束。向透析好的納米膠束加入2.5%的F-68,凍存得到不含葉酸的紫杉醇納米粒。本發明制備的納米粒經透射電鏡觀察,形態規則呈球形,分散好,大小均勻。本實施例制備的不含葉酸的紫杉醇納米粒的透射電鏡見圖1,不含葉酸的紫杉醇納米粒的粒徑分布圖見圖2實施例3:葉酸紫杉醇納米粒的制備稱取聚合物PEG-PLA270mg,稱取聚合物polate-PEG-PLA30mg,使帶葉酸的聚合物比率占總聚合物的10%,溶于1ml四氫呋喃中,配制成300mg/ml的有機溶液。避光稱取紫杉醇50mg,溶于1ml的二氯甲烷中,配成濃度為50mg/ml的有機溶液。將兩者溶液充分混合制成均一的溶液,向該混合溶液中加入3ml聚乙烯醇,30-40W超聲10min,使藥物聚合物溶液充分的混合,將混合好的有機溶液以20-30滴/分鐘的速度滴加到30ml超純水中,冰浴300W超聲20min。將所得的乳液常溫常壓下水浴攪拌4h,揮發有機溶劑,離心(14000r/min)30min,收集沉淀,用蒸餾水洗滌4次,重新水化。將重新水化后的膠束溶液放入透析帶(3500KD)中將透析袋置于水中進行透析,將沒有完全反應的有機溶解和藥物透析出來,即得到葉酸紫杉醇納米膠束。向透析好的納米膠束加入2.5%的F-68,凍存的到含葉酸的紫杉醇納米粒。本發明制備的納米粒經投射電鏡觀察,形態規則呈球形,分散好,大小均勻。將凍干后的紫杉醇納米粒生理鹽水復溶,所得的紫杉醇納米粒復溶后的透射電鏡圖見圖3,復溶后的紫杉醇納米粒的粒徑分布圖見圖4。圖1和圖3分別為凍干前后的透射電鏡圖,對比可以得出,凍存前后紫杉醇納米粒的形狀均為球形,大小均勻,分散佳,凍存對紫杉醇納米粒沒有形態學的改變,但是凍存有利于納米粒的儲存,并且凍存過程中將對機體有害的有機溶劑除去,減少了有機溶劑對機體的損害,為納米粒用于臨床提供了條件。圖2和圖4分別為凍干前后的粒徑分布圖,由兩個圖可以看出,凍干前后粒徑均在100-150nm之間,粒徑沒有發生較大的變化,保留了紫杉醇納米粒特有的納米粒特性。上述雖然結合附圖對本發明的具體實施方式進行了描述,但并非對本發明保護范圍的限制,所屬領域技術人員應該明白,在本發明的技術方案的基礎上,本領域技術人員不需要付出創造性勞動即可做出的各種修改或變形仍在本發明的保護范圍以內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1