一種多肽復合物作為GLP?1藥物載體的應用、方法及其融合蛋白復合物與流程
技術領域
本發明創造屬于生物技術領域,涉及一種基于Titin-Telethoninβ-折疊片結構的多肽復合物作為多肽及蛋白質藥物的載體,延長多肽或者蛋白質類藥物半衰期的應用及方法,同時經過這種多肽復合物對多肽或者蛋白質類藥物的設計和改造,獲得了具有藥用作用的融合蛋白復合物。
背景技術:
目前,絕大多數肽類和其類似物藥物為激素或具有激素作用的肽衍生物,具有生物活性和特異性較高,可溶性強,低毒性等優點,現已成為藥物研發的熱點領域。肽類或蛋白質類藥物雖然具有較強的生物學活性,但在體內發揮藥理學作用時,也具有以下限制因素:1)口服利用度低,給藥方式通常選用注射;2)親水性強,不易通過生理屏障;3)較高的構象靈活性,有時與受體和靶標進行結合時缺乏特異選擇性,易導致副作用。除以上的因素,還有一個最為主要的限制因素為其在體內半衰期較短。一些肽或蛋白質類藥物的體內半衰期僅有幾分鐘,從而導致到達靶組織的藥物濃度過低,不能發揮足夠的藥效,嚴重限制了臨床應用。
半衰期較短一般有兩方面原因造成,一是在血漿、肝臟和腎臟中存在著大量的肽酶(Peptidase)和蛋白酶(Protease),肽或蛋白質在酶的作用下迅速降解;二是在血漿中分子量較小的肽類或蛋白質類藥物(低于50kDa),由于腎臟的過濾作用而被快速排出。延長肽類或蛋白質類藥物的半衰期,不但可以改善藥物的藥代動力學和藥效學特征,而且可以減少用藥量和用藥次數,而這也是現在肽類或蛋白質類藥物臨床應用上的難題。
目前,已經開發出一系列的延長肽類或蛋白質類藥物半衰期的方法,如提高肽類或蛋白質類藥物對酶的穩定性來減少酶的降解;或者通過增加肽類或蛋白質類藥物的流體力學體積(Hydrodynamic Volume)和FcRn介導的再生作用來減少腎臟過濾排出。另外,將較小的多肽或者蛋白質類藥物與大分子物質通過基因融合或者化學偶聯的方式連接在一起,不僅可以增大其分子量,使其超過腎臟過濾的臨界值(40-50kDa),減少腎臟的過濾排出;而且可以依靠大分子量物質的空間位阻效應,減少蛋白質酶等對多肽或者蛋白質藥物的降解,從而極大的延長多肽或者蛋白質類藥物的半衰期,增加了臨床應用的潛力。
目前,應用的比較成熟的大分子量載體有PEG,Albumin和IgG-Fc等。現在最普遍用的一種方法,就是通過化學交聯的方式將不同分子量的PEG與多肽或蛋白質類藥物的特異位點相連,能顯著延長多肽或蛋白質藥物的半衰期。PEG修飾增加蛋白質和多肽藥物的可溶性,提高了穩定性,降低免疫原性,減少蛋白酶的降解,延長在血液中的滯留時間,延長了藥物的作用時間。但是,PEG修飾也存在一些問題,如生產成本較高;有些PEG修飾的藥物其活性降低;近年來又發現PEG修飾的蛋白和其降解物容易在腎臟中累積,干擾了正常腎臟的過濾作用;人和動物能夠對PEG產生一定的抗體,表明其具有一定的免疫原性。
因此,有必要開發一種免疫原性更低或無免疫原性,在體內更穩定的肽或者蛋白質類藥物的載體,使多肽或者蛋白質類藥物既能保持原來的活性,其半衰期又得到延長,從而更好的應用在臨床上。
肌原纖維是由肌節(sarcomere)構成的,Z-盤(Z-disk)是肌節的分界線。在Z-盤中錨定著許許多多的蛋白質,構成了細胞的骨架成分,起著維持細胞結構和肌肉伸縮的重要作用。在Z-盤中,titin分子起著維持肌節完整性的重要作用。Titin分子是自然界中已知的最大的蛋白質,其分子量約為4MDa,長度約為1μm,其N端錨定在Z-盤中,其C端則跨越了肌節一半的長度到達M線。電鏡圖像和基因序列分析表明,titin分子是由300多個免疫球蛋白樣結構域(Immunoglobulin-like,簡寫為Ig)和纖維連接蛋白III(fibronectin-like,簡寫為FN-III)樣的結構域,以及其他一些結構域(如PEVK結構域)等構成。
在Z-盤中,titin的N端與許多蛋白質分子能發生相互作用,這些相互作用能在結構上支持肌原纖維的功能。在這些蛋白質分子中有一種特殊的分子,Telethonin,其能將兩個titin分子的N端通過非共價鍵連接在一起。
晶體結構分析表明,Telethonin N端(TeN)的偽對稱結構(pseudosymmetric structure)通過氫鍵等非共價鍵將titin分子的N端的兩個Ig結構域(Z1Z2)以反向平行的方式連接起來,構成了獨特的β-折疊片結構,這種結構類型在先前的研究中僅在蛋白質-DNA復合物中被發現(Zou PJ et al,Nature,12Jan.2006,439:229-233)。這種交聯代表著一種新型的配體和受體相互作用的方式。傳統的配體和受體的相互作用是配體分子插入到受體分子的結合結構域中,而Telethonin與Z1Z2的相互作用是配體分子Telethonin的N端通過β-折疊片的交聯作用將兩個titin分子的N端(Z1Z2)結合在一起。這種相互作用對于穩定titin的N端具有非常重要的作用。
研究中,常將包含這種titin-Telethoninβ-折疊片結構的復合物稱為Z1Z2/Telethonin復合物。Steered molecular dynamics(SMD)模擬顯示,這種Z1Z2/Telethonin復合物所能承受的機械力遠遠大于單獨的Z1Z2分子(Lee EH,et al,Structure,Mar.2006,14(3):497-509)。而單分子機械力實驗表明,Z1Z2/Telethonin復合物能夠承受700pN的拉力(Bertz M,et al,Proc Natl Acad Sci USA,11Aug.2009,106(32):13307-133310),超過目前已知的蛋白質分子或者復合物所能承受的拉力,因此Z1Z2/Telethonin復合物是非常穩定的一種結構。
雖然對Z1Z2/Telethonin復合物這種獨特的結構已經取得了一定的研究進展,但對于這種結構在各方面的應用卻尚未見報道,而擁有這種結構的復合物在作為藥物載體方面的適用性的研究至今也更未涉及。
技術實現要素:
本發明創造提供一種基于Titin-Telethoninβ-折疊片結構的多肽復合物作為多肽或蛋白質藥物載體的應用,所述多肽復合物含有:(i)多肽A:包含titin分子N端的兩個Ig結構域(Z1Z2)的多肽、其同源物、類似物或衍生物;(ii)多肽B:包含Telethonin分子N端的β-折疊片區域的多肽、其同源物、類似物或衍生物;所述多肽B將兩個所述多肽A結合在一起構成β-折疊片結構。
進一步,所述多肽復合物中,所述多肽A為包含SEQ ID NO:6中31-221位所示的氨基酸序列的片段、其保守性變體、生物活性片段或衍生物,所述多肽B為包含SEQ ID NO:2中30-116位所示的氨基酸序列的片段、其保守性變體、生物活性片段或衍生物,且所述多肽B將兩個所述多肽A結合在一起構成β-折疊片結構。
在一些方案中,所述多肽復合物中所述多肽A為包含與SEQ ID NO:6中31-221位所示的氨基酸序列至少具有60%相同性的序列的多肽片段,優選為至少具有70%相同性,還優選為至少具有80%相同性,再優選為至少具有90%相同性,更優選為至少具有95%、96%、97%、98%、99%的相同性;所述多肽B為包含與SEQ ID NO:2中30-116位所示的氨基酸序列至少具有60%相同性的序列的多肽片段,優選為至少具有70%相同性,還優選為至少具有80%相同性,再優選為至少具有90%相同性,更優選為至少具有95%、96%、97%、98%、99%的相同性。
在一些方案中,所述多肽復合物中所述多肽A包含從SEQ ID NO:6中31-221位所示的氨基酸序列中連續或間斷地缺失、替換或插入0-36中任意數值個氨基酸殘基得到的多肽之一,優選為0-20中任意數值個,更優選為0-10中任意數值個,最優選為0、1、2、3、4、5、6個;所述多肽B包含從SEQ ID NO:2中30-116位所示的氨基酸序列中連續或間斷地缺失、替換或插入0-36中任意數值個氨基酸殘基得到的多肽之一,優選為0-20中任意數值個,更優選為0-10中任意數值個,最優選為0、1、2、3、4、5、6、7、8個。
進一步,所述多肽A和/或多肽B還可以包含或連接蛋白質純化標識序列,所述蛋白質純化標識序列可選自HA、GST、His6、MBP、Flag、Halo、CBD、Srep中的一種或多種。
進一步,所述多肽A和/或多肽B還可以包含或連接一段或多段蛋白質間隔或連接序列(linker),所述蛋白質間隔或連接序列用于將外源的氨基酸序列插入多肽A和/或多肽B的氨基酸序列中,而不破壞所述多肽復合物的β-折疊片結構。
優選的,所述多肽復合物中,所述多肽A為具有SEQ ID NO:6中31-221位所示的氨基酸序列,多肽B為具有SEQ ID NO:2中30-116位所示的氨基酸序列或將其Cys突變為Ser的變體序列(SEQ ID NO:4中30-116位所示的氨基酸序列)。
還優選的,所述多肽復合物中,所述多肽A為具有SEQ ID NO:6中27-221位所示的氨基酸序列,多肽B為具有SEQ ID NO:2中27-116位所示的氨基酸序列或具有SEQ ID NO:4中27-116位所示的氨基酸序列。
更優選的,所述多肽復合物中,所述多肽A為具有SEQ ID NO:6中25-221位所示的氨基酸序列,多肽B為具有SEQ ID NO:2中25-116位所示的氨基酸序列或具有SEQ ID NO:4中25-116位所示的氨基酸序列。
最優選的,所述多肽復合物中,所述多肽A為具有SEQ ID NO:6所示的氨基酸序列,多肽B為具有SEQ ID NO:2所示的氨基酸序列或具有SEQ ID NO:4所示的氨基酸序列。
進一步,所述多肽復合物包含編碼所述多肽A的核酸序列A及編碼所述多肽B的核酸序列B;所述核酸序列A選自下述的一種核酸序列或其變體:(a)編碼具有SEQ ID NO:6中31-221位氨基酸序列的多肽、其保守性變體、生物活性片段或衍生物的核酸序列;(b)與核酸序列(a)互補的核酸序列;(c)與核酸序列(a)或(b)具有至少70%相同性的核酸序列;所述核酸序列B選自下述的一種核酸序列或其變體:(d)編碼具有SEQ ID NO:2中30-116位或SEQ ID NO:4中30-116位氨基酸序列的多肽、其保守性變體、生物活性片段或衍生物的核酸序列;(e)與核酸序列(d)互補的核酸序列;(f)與核酸序列(d)或(e)具有至少70%相同性的核酸序列;且所述核酸序列B編碼的多肽B能夠將所述核酸序列A編碼的兩個多肽A結合在一起構成β-折疊片結構。
編碼所述多肽A的核酸序列A及編碼所述多肽B的核酸序列B可以分別優選為根據待構建表達載體的生物體的密碼子偏愛性設計的核酸序列,例如根據原核生物大腸桿菌偏愛的密碼子設計的分別編碼所述多肽A及多肽B的核酸序列。
編碼所述多肽A的核酸序列A還可以優選為具有SEQ ID NO:5中91-663位的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體,還優選為具有SEQ ID NO:5中79-663位的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體,更優選為具有SEQ ID NO:5中73-663位的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體,最優選為具有SEQ ID NO:5的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體;編碼所述多肽B的核酸序列B還可以優選為具有SEQ ID NO:1中88-348位或SEQ ID NO:3中88-348位的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體,還優選為具有SEQ ID NO:1中79-348位或SEQ ID NO:3中79-348位的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體,更優選為具有SEQ ID NO:1中73-348位或SEQ ID NO:3中73-348位的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體,最優選為具有SEQ ID NO:1或SEQ ID NO:3的核苷酸序列、其保守性變體、互補序列或互補序列的保守性變體。
本說明書和權利要求書中使用的下列術語,除非另外說明具有下述一般含義,且下述含義被認為在本領域技術人員的知識范圍之內:
“核酸序列”指寡核苷酸、核苷酸或多核苷酸及其片段或部分,也可以指基因組或合成的DNA或RNA,可以為單鏈或雙鏈,可以為有義鏈或反義鏈。
“多肽”泛指具有氨基酸序列的肽鏈,包括寡肽、肽、狹義的多肽(20個氨基酸殘基以上的肽)或蛋白質序列及其片段或部分,可以使糖基化或非糖基化的,氨基酸序列中一個或多個氨基酸殘基可以被修飾或未被修飾。當其氨基酸序列涉及一種天然存在的蛋白質分子的氨基酸序列時,這種“多肽”或“蛋白質”不意味著將氨基酸序列限制為與該天然存在的蛋白質分子相關的完整的或完全一致的天然氨基酸或其序列。
“同源”包括完全同源和部分同源,在描述多肽、蛋白質或氨基酸序列時,指具有相同或相似的結構或功能,或具有相似的氨基酸序列;在描述核酸序列時,指具有相似性或互補的核酸序列;“同源”在本發明具有相對較為廣泛的含義,例如,包括具有一定百分比的相同性的序列(氨基酸序列或核酸序列),或包括序列的變體。
“類似物”指基本上保持本發明titin分子N端的兩個Ig結構域(Z1Z2)的多肽A和Telethonin分子N端β-折疊片區域的多肽B的結構或生物學功能或活性的多肽,只要能夠基本形成二者間β-折疊片結構。本發明的多肽“類似物”可以包括:(I)在序列中連續或間隔地插入、缺失、替換一個或多個氨基酸殘基,且所述一個或多個氨基酸殘基的插入、缺失、替換在同一序列中可同時或不同時存在;(II)序列中一個或多個氨基酸殘基的基團被其他基團替換或缺失;(III)序列中一個或多個氨基酸殘基被修飾。
“衍生物”在描述多肽、蛋白質或氨基酸序列時,指由原多肽、蛋白質或氨基酸序列衍生而來的關聯多肽、蛋白質或氨基酸序列,本發明的多肽復合物在能夠基本形成β-折疊片結構時包括這樣的衍生物,例如,本發明中多肽A或多肽B包括這樣的衍生物:(IV)成熟多肽與另一種化合物融合,或者(V)在氨基酸序列中融合或插入附加的氨基酸序列(linker、蛋白質純化標識序列、酶切位點等);在描述核酸序列時,指由原始序列衍生未來的關聯序列,可以包括:(VI)序列或基因中連續或間隔地插入、缺失、替換一個或多個堿基(優選為等位基因的替換),且所述一個或多個氨基酸殘基的插入、缺失、替換在同一序列或基因中可同時或不同時存在;(VII)序列或基因中一個或多個堿基被修飾;(VIII)序列或基因中融合或插入編碼附加的氨基酸序列的基因。
“保守”指所涉及的氨基酸序列或核酸序列與原始序列具有較高的相似性或同一性,能夠維持其原始序列基本的結構、生物學活性或功能,一般可以通過相似的氨基酸殘基替換或等位基因(簡并密碼子)替換等獲得,例如,賴氨酸和精氨酸、亮氨酸和異亮氨酸之間的替換等。
“變體”指具有一個或多個氨基酸或核苷酸改變的氨基酸序列或核酸序列,所述改變可包括氨基酸序列或核酸序列中氨基酸或核苷酸的插入、缺失或替換。變體可具有保守性改變,其中替換的氨基酸與原氨基酸具有相似的結構或化學性質,如亮氨酸和異亮氨酸之間的替換,也可具有非保守性改變。值得指出的是,本發明中非保守性改變的變體也能夠達到甚至超越天然氨基酸序列構成的多肽復合物所具有的性質及能夠達到的效果,例如,研究發現,將SEQ ID NO:2所示的氨基酸序列中半胱氨酸全部替換為絲氨酸,能夠去除多肽B鏈中β-折疊片區域多余的二硫鍵,使得本發明的多肽復合物的β-折疊片結構更加穩定。
“相似性”、“相似”、“相同性”或“同一性”在用于描述多肽或蛋白質結構、氨基酸序列或核酸序列時,指其包含的序列之間排列對比時相應位置氨基酸殘基或核苷酸相同的程度,可以用百分數表示。
本發明還提供了上述多肽復合物作為藥物載體的應用的方法,所述方法包括:將一個或多個多肽或蛋白質類藥物的編碼序列插入或連接所述多肽A和/或多肽B的編碼序列的一個或多個合適位點,獲得融合基因,并使融合基因在表達系統中表達獲得含有該多肽或蛋白質藥物的多肽A和/或多肽B的融合蛋白,然后使融合蛋白和其配合物接觸,構成具有藥用作用的融合蛋白復合物。其中,所述配合物是指能夠與所述融合蛋白形成β-折疊片結構的多肽A或多肽B或另一連接了多肽或蛋白質藥物的多肽A或多肽B的融合蛋白。
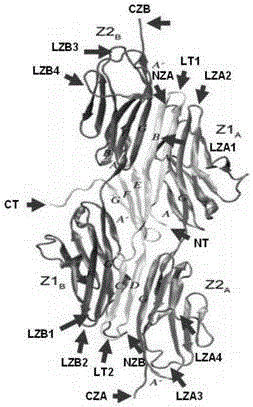
其中,所述位點是多肽復合物中能夠與多肽或蛋白質藥物結合的區域,本發明中優選的位點包括下述16個(圖1):多肽B的N端(NT)、C端(CT)和內部的兩個loop區(LT1、LT2),兩個多肽A的N端(NZA、NZB)、C端(CZA、CZB)和八個loop區(LZA1、LZA2、LZA3、LZA4、LZB1、LZB2、LZB3、LZB4)。
其中,多肽或蛋白質藥物能夠在上述10個loop區中的任意位置與所述多肽復合物結合。
進一步,具體至具有SEQ ID NO:2、SEQ ID NO:4和SEQ ID NO:6的氨基酸序列的多肽A和多肽B的中,上述10個loop區的位置分別為:
多肽B內部的兩個loop區:SEQ ID NO:2或SEQ ID NO:4中第42-43位和第72-73位氨基酸序列;
多肽A內部的四個loop區:SEQ ID NO:6中第57-62位、第111-113位、第142-147位、第170-175位氨基酸序列。其中,所述融合基因還包括蛋白質純化標識序列、酶切位點、各種柔性或剛性linker的編碼序列、調控序列等。所述融合基因可以通過PCR、酶切、酶連、基因重組、全基因合成等方式獲得。所述表達系統可以采用大腸桿菌表達系統(可選用pET系列質粒做表達載體)、酵母表達系統(如畢赤酵母)、昆蟲表達系統(如家蠶型多角體病毒)、哺乳動物表達系統或植物表達系統等,優選為采用pET系列質粒做表達載體的大腸桿菌表達系統。
進一步,所述方法還包括融合基因獲得后的測序步驟以及融合蛋白的純化步驟。
本發明還提供了上述多肽復合物作為藥物載體的應用的另一種方法,包括用一些偶聯試劑將多肽或者蛋白質藥物與多肽復合物的一個或多個位點進行連接。如使用碳化二亞胺、戊二醛和二異氰酸化合物等常用偶聯劑將多肽或蛋白質類藥物與多肽A和/或多肽B的N端或C端進行連接,然后使連接后的多肽與其配合物接觸,獲得具有藥用作用的融合蛋白復合物;或者通過基因突變的方式在多肽A和/或多肽B內部引入-SH,然后利用馬來酰亞胺等偶聯劑與多肽或蛋白藥物相連,然后使連接后的多肽與其配合物接觸,獲得具有藥用作用的融合蛋白復合物。
本發明的另一目的是提供一種延長藥物半衰期的方法,所述方法使用一種多肽復合物作為載體,所述多肽復合物含有:(i)多肽A:包含titin分子N端的兩個Ig結構域(Z1Z2)的多肽、其同源物、類似物或衍生物;(ii)多肽B:包含Telethonin分子N端的β-折疊片區域的多肽、其同源物、類似物或衍生物;所述多肽B將兩個所述多肽A結合在一起構成β-折疊片結構。
進一步,所述方法包括:將一個或多個多肽或蛋白質藥物的編碼序列插入或連接所述多肽A和/或多肽B的編碼序列的一個或多個合適位點,獲得融合基因,并使融合基因在表達系統中表達獲得含有該多肽或蛋白質藥物的多肽A和/或多肽B的融合蛋白,然后使融合蛋白和其配合物接觸,構成具有藥用作用的融合蛋白復合物。其中,所述配合物是指能夠與所述融合蛋白形成β-折疊片結構的多肽A或多肽B或另一連接了多肽或蛋白質藥物的多肽A或多肽B的融合蛋白。
其中,所述位點是所述多肽復合物中能夠與多肽或蛋白質藥物結合的區域,本發明中優選的位點包括下述16個(圖1):多肽B的N端(NT)、C端(CT)和內部的兩個loop區(LT1、LT2),兩個多肽A的N端(NZA、NZB)、C端(CZA、CZB)和八個loop區(LZA1、LZA2、LZA3、LZA4、LZB1、LZB2、LZB3、LZB4)。
進一步,具體至具有SEQ ID NO:2、SEQ ID NO:4和SEQ ID NO:6的氨基酸序列的多肽A和多肽B的中,上述10個loop區的位置分別為:
多肽B內部的兩個loop區:SEQ ID NO:2或SEQ ID NO:4中第42-43位和第72-73位氨基酸序列;
多肽A內部的四個loop區:SEQ ID NO:6中第57-62位、第111-113位、第142-147位、第170-175位氨基酸序列。其中,所述融合基因還包括蛋白質純化標識序列、酶切位點、各種柔性或剛性linker的編碼序列、調控序列等。所述融合基因可以通過PCR、酶切、酶連、基因重組、全基因合成等方式獲得。所述表達系統可以采用大腸桿菌表達系統(可選用pET系列質粒做表達載體)、酵母表達系統(如畢赤酵母)、昆蟲表達系統(如家蠶型多角體病毒)、哺乳動物表達系統或植物表達系統等。
進一步,所述方法還包括融合基因獲得后的測序步驟以及融合蛋白的純化步驟。
本發明還提供了一種具有藥用作用的融合蛋白復合物,所述融合蛋白復合物包含由多肽或蛋白質藥物與多肽A和/或多肽B構成的融合蛋白,且該融合蛋白與多肽A或多肽B或另外的所述融合蛋白形成β-折疊片結構。
其中,所述多肽A為包含titin分子N端的兩個Ig結構域(Z1Z2)的多肽、其同源物、類似物或衍生物;所述多肽B為包含Telethonin分子N端的β-折疊片區域的多肽、其同源物、類似物或衍生物。
進一步,所述融合蛋白中,多肽或蛋白質藥物位于多肽A和/或多肽B的一個或多個下述位點:多肽B的N端(NT)、C端(CT)和內部的兩個loop區(LT1、LT2),兩個多肽A的N端(NZA、NZB)、C端(CZA、CZB)和八個loop區(LZA1、LZA2、LZA3、LZA4、LZB1、LZB2、LZB3、LZB4)。
進一步,具體至具有SEQ ID NO:2、SEQ ID NO:4和SEQ ID NO:6的氨基酸序列的多肽A和多肽B的中,上述10個loop區的位置分別為:
多肽B內部的兩個loop區:SEQ ID NO:2或SEQ ID NO:4中第42-43位和第72-73位氨基酸序列;
多肽A內部的四個loop區:SEQ ID NO:6中第57-62位、第111-113位、第142-147位、第170-175位氨基酸序列。進一步,所述融合蛋白中,多肽或蛋白質藥物可以有一種或多種。
進一步,所述融合蛋白的編碼序列還可以包括蛋白質純化標識序列、酶切位點、各種柔性或剛性linker的編碼序列、調控序列等。
本發明的多肽復合物能夠承載的藥物,包括在多肽復合物作為藥物載體的應用、方法、延長藥物半衰期的方法、以及具有藥用作用的融合蛋白復合物幾個主題中涉及到的多肽或蛋白質藥物,可以是能夠與下述受體相互作用的多肽或蛋白質藥物,包括這些受體的激活劑(agonist)或拮抗劑(antagonist):(a)含離子通道的受體,如N膽堿受體、興奮性氨基酸受體等;(b)G-蛋白偶聯受體(GPCR);(c)酶活性的受體,如胰島素受體、表皮生長因子受體、生長激素受體、干擾素受體等;(d)細胞核激素受體,如腎上腺皮質激素受體、甲狀腺素受體等。
其中,GPCR是人體內最大的膜受體蛋白家族,現代藥物中約有50%的多肽或蛋白質藥物以GPCR為作用靶點。這些多肽或者蛋白質藥物尤其適合以本發明多肽復合物作為載體,非限定性的例子可以針對以下幾種GPCR的一種或幾種:一,腫瘤相關GPCR,如Samatostatin(SST)/Growth(GH)受體群,Gonadotropin releasing hormone(GnRH)/Luteinizing hormone(LH)/類固醇激素受體群,CXCR4等等;二,免疫系統GPCR,如CC chemokine受體或者CXC chemokine受體,Formyl多肽受體等等;三,內分泌系統GPCR的多肽或者蛋白質藥物,如GLP-1受體,Glucagon受體,NPYY2受體,Canabinoid受體CB1和CB2,Ghrelin受體GHS-R,Melanocortin受體MC4以及一些孤兒受體如GPR35,GPR85,GPR101等等。
進一步,所述多肽復合物能夠承載的藥物還包括經改造后的多肽或蛋白質藥物,例如在保持藥物活性的條件下對多肽或蛋白質藥物的氨基酸序列或其核酸序列進行連續或間隔地插入、缺失、替換、修飾一個或多個氨基酸殘基或堿基,且所述一個或多個氨基酸殘基或堿基的插入、缺失、替換、修飾在同一序列或基因中可同時或不同時存在。所述改造可以在本發明的融合蛋白復合物制備過程的任一步驟進行,例如,在制備多肽或蛋白質藥物與多肽A和/或多肽B的融合蛋白之前先對多肽或蛋白質藥物的編碼序列進行突變,然后再進行基因融合獲得編碼融合蛋白的融合基因;或者先獲得編碼多肽或蛋白質藥物與多肽A和/或多肽B的融合蛋白的融合基因,再對融合基因中多肽或蛋白質藥物的編碼序列部分進行突變等。
本發明說明書和權利要求中,在涉及到除本發明多肽復合物作為藥物載體應用外的其他主題時,諸如多肽復合物作為藥物載體的應用的方法、一種延長藥物半衰期的方法、一種具有藥用作用的融合蛋白復合物等,所提及的多肽A和多肽B,均與本發明在多肽復合物作為藥物載體應用的主題內討論的多肽A和多肽B具有同等的含義和范圍,不再贅述。
本發明的多肽復合物可以抗蛋白酶水解,穩定性較好,其分子量大小在50kDa左右或更高,腎臟不能將其快速濾除;其源自于人體的肌肉蛋白質,由四個類免疫球蛋白的亞基所組成,免疫原性較低;且多肽復合物含有多個位點,在這些位點接入或插入肽或小分子蛋白質而不影響復合物的結構及穩定性;將本發明的多肽復合物作為多肽或蛋白質藥物的載體的同時,能夠快速地對多肽或蛋白質藥物進行設計和改造,構建新型的具有藥用作用的融合蛋白復合物,在保持原有藥物活性的同時,有效延長多肽或者蛋白質類藥物的半衰期,更好地在臨床上得到應用。
附圖說明
圖1是本發明中多肽復合物晶體結構及藥物接入或插入位點示意圖。其中,Z1A和Z2A分別表示多肽復合物中一條多肽鏈上的兩個Ig結構域,Z1B和Z2B分別表示多肽復合物中另一條多肽鏈上的兩個Ig結構域。
圖2是實施例一中Z1Z2/GLTe(A)和Z1Z2/TeN(B)純化后SDS和Native-PAGE。
圖3是實施例一中不同濃度GLP-1和Z1Z2/GLTe刺激RINm5F細胞產生Camp。
圖4是實施例一活性檢測結果圖。其中,(A),一次腹腔注射25nmol Z1Z2/GLTe,Z1Z2/TeN和GLP-1對血糖的控制作用;(B)表示A實驗的曲線下面積(AUC);(C)表示一次腹腔注射25nmol Z1Z2/GLTe和GLP-1后,大鼠血漿中胰島素含量變化;(D)表示一次腹腔注射25nmol Z1Z2/GLTe 24h后,進行OGTT;(E)表示D實驗的AUC;(F)表示不同濃度Z1Z2/GLTe對血糖的控制作用;(G)表示E實驗的AUC。(不同處理組與PBS組進行比較,*表示p<0.05,**表示p<0.01,***表示p<0.001。)
圖5是實施例一Z1Z2/GLTe和GLP-1在大鼠血清中的穩定性測試圖。(**表示p<0.01,***表示p<0.001。)
圖6是實施例一Z1Z2/GLTe和GLP-1通過腹腔注射(A)和靜脈注射(B)大鼠后,活性GLP-1在大鼠血漿中的含量隨時間變化曲線。
圖7是實施例一每天注射一次Z1Z2/GLTe對糖尿病小鼠空腹血糖長期控制的影響曲線。
圖8是實施例一腹腔注射25nmol Z1Z2/GLTe和等體內PBS后,24h內對STZ誘導的糖尿病模型小鼠(A)和正常小鼠(B)飲食量的影響對比圖。(**表示p<0.01,***表示p<0.001。)
圖9是實施例二活性檢測結果圖。其中,(A)表示不同濃度(1,5,25nmol/kg)Z1Z2/GLTe-G進行OGTT實驗結果圖;(B)表示不同濃度(1,5,25nmol/kg)Z1Z2/GLTe-G刺激胰島素分泌實驗結果圖。(**表示p<0.01,***表示p<0.001。)
圖10是實施例二中體內穩定性檢測實驗中Z1Z2/GLTe-G經靜脈注射入大鼠后,血漿中GLP-1的含量變化曲線圖。
圖11是實施例三中Z1Z2/SSTe純化后SDS和Native-PAGE。
圖12是實施例三中活性檢測實驗中大鼠體內生長激素含量變化曲線圖。
圖13是實施例三中半衰期檢測實驗中Z1Z2/SSTe經靜脈注射入大鼠后,血漿中SST的含量變化曲線圖。
具體實施方式
下面通過具體實施例對本發明創造進行進一步說明。應理解,這些實施例僅用于說明本發明而不用于限定本發明的范圍。下述實施例中未注明具體條件的實驗方法,通常按照常規條件,或本領域技術人員在知識范圍內能夠推知的條件,或按照制造廠商所建議的條件。下述實施例中涉及的試劑和儀器用具,通常為商用可市購產品,或能夠通過其他公開途徑獲得的產品。
實施例一多肽復合物作為GLP-1的載體
GLP-1(Glucagon-like peptide 1)是一種葡萄糖依賴的促胰島素分泌肽,其具有兩種活性形式,GLP-1(7-36)酰胺和GLP-1(7-37)。GLP-1在體內能與屬于G蛋白偶聯受體(GPCRs)的GLP-1受體結合,能促進胰島β細胞分泌胰島素,抑制胰高血糖素的分泌,控制血糖,是有效治療2型糖尿病的多肽激素,但其在體內受到二肽基肽酶IV(Dipeptidyl Peptidase IV,DPP-IV)的降解,其半衰期僅2min左右,臨床應用時需要高劑量頻繁注射,嚴重限制了其作為藥物在臨床上的應用。
實驗部分:
1.基因融合
將具有編碼Z1Z2的基因序列(如SEQ ID NO:5中73-663位,以下簡稱Z序列)和具有編碼Telethonin N端(TeN)的基因序列(如SEQ ID NO:1中73-348位,以下簡稱T序列)通過NcoI/KpnI酶切位點克隆到一個經過改造的pET24d的載體上(在pET24d上引入以6xHis標簽和一個TEV蛋白酶酶切位點),并且將T序列中的Cys突變為Ser(Zou PJ et al,J Biol Chem.24Jan 2003,278(4):2636-2644),得到pET-Z1Z2(原pET24d載體之外的序列部分如SEQ ID NO:5所示)和pET-TeN(原pET24d載體之外的序列部分如SEQ ID NO:3所示)質粒。
通過PCR的方法將GLP-1(7-37)連接到T序列的N端,中間通過linker(GGGGSGGGGSGGGGS)連接。用引物TeN-F、TeN-R將pET-TeN質粒全長進行擴增,
TeN-F:(SEQ ID NO:13)
GGTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGCCATGGCTACCTCAGAGCTGAG(下劃線部分為linker密碼子)
TeN-R:(SEQ ID NO:14)
CTGAAAATAAAGATTCTCAGTAGTGGGGAT
GLP-1全長基因采用重疊延伸法擴增,引物分別為GLP-1-F、GLP-1-R,
GLP-1-F:(SEQ ID NO:15)
P’CATGCGGAAGGCACCTTTACCAGCGATGTGAGCAGCTATCTGGAAGGCCAGGC
GLP-1-R:(SEQ ID NO:16)
P’GCCACGGCCTTTCACCAGCCACGCAATAAACTCTTTCGCCGCCTGGCCTTCCAGAT
P’表示5’端磷酸化。
先用TeN-F和TeN-R擴增全長且帶有(GGGGS)3的pET-TeN,然后用T4DNA ligase將pET-TeN和帶有5’磷酸的GLP-1基因進行連接,轉化,挑選單克隆提取質粒后測序。測序正確的質粒命名為pET-24d-GLTe,原pET24d載體之外的序列部分如下(SEQ ID NO:7):
其中,箭頭所指為TEV酶酶切位點,雙下劃線為6xHis標簽,單下劃線為GLP-1氨基酸序列,曲線部分為linker。
2.表達
將pET-Z1Z2、pET-TeN和pET-24d-GLTe分別轉化入E.coli BL21(DE3)感受態細胞中,挑取單菌落于含60μg/ml卡那霉素的LB培養基中,37℃搖床過夜培養后,以1%接種量轉接下一級,37℃繼續培養至菌液OD600約為0.4-0.6時加入終濃度為1.0mmol/L的IPTG,并于30℃誘導表達12h。待表達時間結束后在4℃,5000g條件下離心10min,收集菌體。
收集菌體后,用平衡緩沖液(25mM Tris/HCl,300mM NaCl,10mM咪唑,pH 8.0)進行重懸,然后,用細胞高壓破碎儀(廣州聚能生物科技有限公司)對菌體進行破碎,破碎結束后,14000rpm離心30min。
對于Z1Z2,吸取上清到Ni2+-NTA柱子(預先用平衡緩沖液進行平衡)中;利用AKTA purifier 10蛋白純化儀對蛋白分別用漂洗緩沖液(25mM Tris/HCl,300mM NaCl,30mM咪唑,pH 8.0)進行漂洗,然后用洗脫緩沖液(25mM Tris/HCl,300mM NaCl,400mM咪唑,pH 8.0)進行洗脫,收集洗脫液。
對于TeN和GLTe則將上清棄掉,用含有8M尿素的平衡緩沖液進行重懸,14000rpm離心30min后,吸取上清加入到用含有8M尿素的平衡緩沖液平衡好Ni2+-NTA柱中;利用AKTA purifier 10蛋白純化儀對蛋白分別用漂洗緩沖液(8M尿素,25mM Tris/HCl,300mM NaCl,30mM咪唑,pH 8.0)進行漂洗,然后用洗脫緩沖液(8M尿素,25mM Tris/HCl,300mM NaCl,400mM咪唑,pH 8.0)進行洗脫,收集洗脫液。
3.純化
分別將等體積TeN和GLTe逐滴加入到Z1Z2中,邊加邊搖動,使最終尿素的濃度達4M;然后將混合溶液在透析液I(25mM Tris/HCl,300mM,pH 8.0)中透析4h,然后換到透析液II(25mM Tris/HCl,pH 8.0)中透析過夜。最終分別形成Z1Z2/TeN復合物和Z1Z2/GLTe復合物。
在Z1Z2/TeN復合物和Z1Z2/GLTe復合物中按1/50的體積比加入TEV蛋白酶,以及TEV蛋白酶酶緩沖液(50mM Tris/HCl pH 8.0,0.5mM EDTA,1mM DTT),在室溫反應2h。
將用TEV蛋白酶酶切后的Z1Z2/TeN復合物和Z1Z2/GLTe復合物過Ni2+-NTA柱,沒有被切完全的或者一些雜蛋白與Ni2+重新結合。流穿為沒有His-tag的Z1Z2/TeN復合物和Z1Z2/GLTe復合物。
用HiTrapTM Q離子交換柱對Z1Z2/TeN和Z1Z2/GLTe進行進一步的純化。分別將Z1Z2/TeN和Z1Z2/GLTe與Q離子交換柱進行結合,然后用AKTA Purifier10蛋白純化系統進行線性洗脫,分別得到純度較高的Z1Z2/TeN復合物和Z1Z2/GLTe復合物。
4.除內毒素
用大腸桿菌表達的蛋白質中含有大量的內毒素,而內毒素會對后期的細胞和動物實驗產生嚴重影響,因此必須去除。用Q離子交換柱純化后的Z1Z2/TeN復合物和Z1Z2/GLTe復合物加入到內毒素去除柱子中(ToxinEraserTM Endotoxin Removal kit,Genscript,Nanjing,China)以除去內毒素,并用內毒素檢測試劑盒(ToxinSensorTM Chromogenic LAL Endotoxin Assay Kit,Genscript)進行內毒素含量檢測,最終Z1Z2/TeN復合物和Z1Z2/GLTe復合物中內毒素的含量小于2EU/ml。
5.GLP-1受體(GLP-1 receptor,GLP-1R)激活實驗
大鼠胰島素瘤細胞RINm5F在含有10%FBS(Life Technology)的RPMI 1640培養基(Life Technology),并放于37℃,5%CO2培養箱中培養。5000個RINm5F細胞接種到96孔細胞培養板中,過夜培養;用不含血清的RPMI 1640培養基清洗兩遍后,將不同濃度的Z1Z2/GLTe復合物和GLP-1(用不含血清的培養基稀釋,并加100μM的IBMX)分別與RINm5F細胞在37℃下孵育20min;然后,裂解細胞,細胞內cAMP的含量按照cAMP-GloTM Assay試劑盒(Promega)說明書進行檢測。
6.活性檢測
為了確定Z1Z2/GLTe對葡萄糖的控制作用,我們進行了口服葡萄糖耐受性實驗(Oral glucose tolerance test,OGTT)。將250g左右大鼠隨機分為4組(n=5),分別為Z1Z2/TeN組,Z1Z2/GLTe組,GLP-1組和PBS組;前3組分別按體重給腹腔注射25nmol/kg,PBS組給予相同體積的PBS,并用血糖儀測血糖濃度。30min后,通過灌胃的方式給予每kg體重2g葡萄糖,此時計為0min,此后分別在10,30,60,90,120min時測血糖。曲線下面積(Area of under curve,AUC)用Graphpad Prism 6.0軟件進行計算。在這個過程中,分別在0,10,30min時通過尾靜脈取血,加入到含有EDTA-Na2的離心管中,4000rpm離心10min,取上清,用Rat/Mouse Insulin Elisa kit(Millipore)測定其胰島素含量。
為了進一步確定Z1Z2/GLTe的濃度與血糖控制的關系,我們進行了不同濃度Z1Z2/GLTe的OGTT實驗;將250g左右SD大鼠隨機分為4組(n=5),為別為1nmol/kg組,5nmol/kg組,25nmol/kg組和PBS組;通過腹腔注射分別將上述濃度劑量的Z1Z2/GLTe和PBS注射入大鼠體內,30min后通過灌胃的方式給予每kg體重2g的葡萄糖,此時計為0min,此后分別在10,30,60,90,120min時用血糖儀測血糖,并分別計算AUC。
為確定Z1Z2/GLTe可以在多長時間內發揮控制血糖的作用,我們進行了腹腔注射藥物24h后對SD大鼠進行OGTT。正常進食的大鼠隨機分為兩組(n=5),分別是PBS組和Z1Z2/GLTe組。Z1Z2/GLTe組,將25nmol/kg的Z1Z2/GLTe通過腹腔注射入大鼠體內,另一組注射等體積的PBS。讓其自由進食12h,然后禁食12h,禁食后進行OGTT。
7.體外穩定性檢測
將Z1Z2/GLTe復合物和GLP-1分別加入新鮮采集的大鼠血清中,使其終濃度為1nM,然后分別在0,0.25,0.5,1,2,4,6,10h進行取樣,取樣后馬上加入DPP-4抑制劑(Millipore)使抑制劑的終濃度為50μM。樣品中活性GLP-1的濃度用Active GLP-1Elisa kit(Millipore)進行檢測。
8.體內穩定性檢測
將Z1Z2/GLTe和GLP-1分別通過腹腔(4nmol)和靜脈(1nmol)注射入SD大鼠體內,分別在0,0.5,1,1.5,2,4,6,10h從尾部取血,滴入預先用EDTA-Na2處理過的Ep管中,取血后立即(30s以內)加入DPP-4抑制劑。樣品中活性GLP-1的濃度用Active GLP-1Elisa kit(Millipore)進行檢測。
9.對糖尿病小鼠血糖及飲食控制
將鏈脲佐菌素(STZ)按45mg/kg體重腹腔注射入KM小鼠中,連續注射5d,然后正常飼喂10d。然后,過夜禁食(12-16h)后測血糖,空腹血糖值>11.1mM的可判斷為糖尿病模型。實驗分為三組,正常組(即非糖尿病模型組);糖尿病小鼠對照組(注射PBS)和糖尿病小鼠實驗組(注射Z1Z2/GLTe),每組6只,分3籠飼養。實驗組每天上午9點按體重給予腹腔注射25nmol/kg Z1Z2/GLTe,對照組和正常組分別給予腹腔注射PBS。每4d過夜禁食(12-16h)一次,第4d測空腹血糖值。
為了研究Z1Z2/GLTe對正常小鼠和糖尿病小鼠飲食的影響。我們將正常KM小鼠(非糖尿病模型)12只,分2組,對照組(PBS)和Z1Z2/GLTe組;糖尿病模型小鼠12只,分2組,對照組(PBS)和Z1Z2/GLTe組。過夜禁食(12-16h)后,正常小鼠和糖尿病小鼠分別稱重,然后Z1Z2/GLTe組分別按重量腹腔注射25nmol/kg Z1Z2/GLTe,PBS組分別腹腔注射同體積的PBS;給予預先稱重的鼠糧,然后分別在2h,4h,8h和24h時稱鼠糧的重量,以鼠糧的減少量除以每籠小鼠的體重為指標測定小鼠的進食量。
結果與分析部分:
1.Z1Z2/GLTe表達純化
從圖2可以看出,經過一系列的純化后,Z1Z2/GLTe在SDS-PAGE圖上表現為兩條非常純的條帶,分別為Z1Z2(上)和GLTe(下);而在Native-PAGE圖上則表現為一條非常純的條帶,為Z1Z2/GLTe復合物。這表明,我們獲得了純度較高的用于后續試驗的Z1Z2/GLTe復合物。
Z1Z2/GLTe復合物經內毒素去除預裝柱處理后,其內毒素含量控制在2EU/ml的范圍內,可以進行后續的細胞和動物實驗。
2.激活GLP-1R實驗
GLP-1R是一個細胞表面表達的G蛋白偶聯受體,GLP-1能夠與細胞表面表達的GLP-1R結合,從而激活下游的信號通路,產生cAMP。通過產生的cAMP的量可以反映與受體的結合情況。結果表明,Z1Z2/GLTe和GLP-1均能與GLP-1R結合,且能以濃度依賴的方式刺激cAMP的產生。Z1Z2/GLTe和GLP-1的EC50值分別為0.35±0.05nM和0.24±0.03nM(圖3)。這表明Z1Z2/GLTe能夠與GLP-1R結合且能強有力的激活GLP-1R,是GLP-1R的一種激動劑。
3.活性檢測
GLP-1在體內能促進胰島β細胞分泌胰島素,抑制胰高血糖素的分泌,控制血糖。結果表明Z1Z2/GLTe能夠非常顯著地降低大鼠血糖。當口服葡萄糖后,血糖含量急劇升高,GLP-1和Z1Z2/GLTe能顯著降低血糖,但是GLP-1只能在30min內發揮作用,而Z1Z2/GLTe卻能在整個120min內有效控制血糖(圖4A,B)。從胰島素的產生來看,Z1Z2/GLTe和GLP-1都能顯著地增加血液中胰島素的含量,因此能夠有效的發揮降血糖的作用(圖4C)。
Z1Z2/GLTe控制血糖的能力具有濃度依賴性,隨著濃度的升高而增加,當Z1Z2/GLTe的注射量為1nmol/kg時,即能顯著降低血糖;當注射量為25nmol/kg時,其抑制血糖升高的能力最強(圖4F,G)。
通過腹腔注射Z1Z2/GLTe 24h后,進行OGTT,發現Z1Z2/GLTe仍然能在60min內控制血糖,表明其在24h后血液中仍然存在Z1Z2/GLTe,且其濃度仍然能夠發揮作用(圖4D,E)。
4.體外穩定性檢測
檢測結果如圖5所示,實驗表明,在大鼠血清中,GLP-1具有較短的半衰期,大約為0.5h,這是由于血清中含有DPP-IV,DPP-IV會將GLP-1N端的兩個氨基酸His和Ala切下,形成沒有活性的多肽;而Z1Z2/GLTe可能由于空間位阻的原因減少了DPP-IV對其的酶切作用,明顯延長了GLP-1的半衰期,其半衰期大約為4h。
5.體內穩定性檢測
實驗數據表明,Z1Z2/GLTe不論通過腹腔注射(圖6A)或靜脈注射(圖6B)都能顯著延長GLP-1半衰期。Z1Z2/GLTe腹腔注射后2h時,血漿中濃度達到最大,而GLP-1則是0.5h時達到最大;然后,GLP-1迅速下降,1h時達到檢測下限,而腹腔注射Z1Z2/GLTe 24h后仍然能檢測到200pM以上的濃度。這表明,Z1Z2/GLTe顯著增加了GLP-1的穩定性,延長了其半衰期,也就增加了其發揮作用的時間。靜脈注射后,Z1Z2/GLTe濃度緩慢下降,而GLP-1迅速下降,0.5h時達到檢測下限。GLP-1經靜脈注射半衰期為1-2min(Kim BJ et al,J Pharmacol Exp Ther,2012,334(3):682-692),而本實驗中Z1Z2/GLTe為67±3.3min,顯著延長了GLP-1的半衰期。
6.Z1Z2/GLTe對糖尿病小鼠血糖的長期控制
Z1Z2/GLTe較原始GLP-1具有更長的半衰期,但是其活性并沒有減弱,因此比GLP-1具有更長時間的控制血糖的能力。通過對糖尿病小鼠血糖控制實驗可以看出(圖7),每天腹腔注射25nmol/kg的Z1Z2/GLTe,能有效降低糖尿病小鼠空腹血糖水平。每天一次注射,能有效地長時間控制空腹血糖水平在一個較低的范圍內。當停止注射(28d)后,其Z1Z2/GLTe組血糖值又開始升高。
7.對進食量的影響
實驗數據表明Z1Z2/GLTe能夠顯著減少糖尿病小鼠(圖8A)和正常小鼠(圖8B)的進食量,這應該是由于Z1Z2/GLTe中的GLP-1能夠增加飽腹感,減少胃排空的原因。減少了進食量也就減少了血糖的進一步升高,對血糖的控制具有積極的作用。
實施例二多肽復合物作為GLP-1(8G)的載體
GLP-1(7-37)由于其氨基酸序列的前兩個氨基酸為HA,所以在血液中易受到DPP-4的切割作用而失去活性,所以將GLP-1(7-37)氨基酸序列中第8位氨基酸突變為Gly,可以有效的抑制DPP-4對其的酶切,從而更長時間延長半衰期。
實驗部分:
1.載體構建
采用下列引物對實施例一中的載體pET-24d-GLTe進行定點突變,將GLP-1(7-37)氨基酸序列中第8位氨基酸Ala突變為Gly。
8A-G-F:(SEQ ID NO:17)
AATCTTTATTTTCAGCATGGCGAAGGCACCTTTACC(下劃線部分為突變位點)
8A-G-R:(SEQ ID NO:18)
GCCATGCTGAAAATAAAGATTCTCAGTAGTGGGGATGTC
測序成功后,命名為pET-24d-GLTe-G,原pET24d載體之外的序列部分如下(SEQ ID NO:9):
其中,箭頭所指為TEV酶酶切位點,雙下劃線為6xHis標簽,單下劃線為GLP-1氨基酸序列,曲線部分為linker,黑斜體部分為突變的氨基酸。
2.表達、純化、除內毒素
采用同實施例1的方法步驟進行,分別獲得純度較高且內毒素的含量小于2EU/ml的Z1Z2/GLTe-G復合物,其他復合物如Z1Z2/TeN復合物可直接由實施例1中的載體或其表達產物或除內毒素后的產物獲得。
3.活性檢測
為了確定Z1Z2/GLTe-G能否有效控制血糖,以及不同濃度與血糖控制的關系,我們進行了不同濃度Z1Z2/GLTe-G的OGTT實驗;將250g左右SD大鼠隨機分為4組(n=5),為別為1nmol/kg組,5nmol/kg組,25nmol/kg組和PBS組;通過腹腔注射分別將上述濃度劑量的Z1Z2/GLTe-G和PBS注射入大鼠體內,30min后通過灌胃的方式給予每kg體重2g的葡萄糖,此時計為0min,此后分別在10,30,60,90,120min時用血糖儀測血糖。
在每組進行OGTT實驗中,10-30min分別從大鼠尾靜脈取血,滴入含有EDTA-Na2的Ep管中,取血漿,用Insulin Elisa kit(Millipore)進行胰島素含量測定。。
4.體內穩定性檢測
將1nmol Z1Z2/GLTe-G通過大鼠尾靜脈注射入大鼠體內,此時計為0h,然后分別在1,2,4,8,24h取血,滴入預先用EDTA-Na2處理過的Ep管中。樣品中活性GLP-1的濃度用GLP-1(Total)Elisa kit(Millipore)進行檢測。
結果與分析部分:
1.活性檢測
對不同濃度Z1Z2/GLTe-G復合物進行OGTT實驗,發現其能劑量依賴性的降低血糖(圖9A)。即使在1nmol/kg時仍然能夠發揮降血糖作用,而25nmol/kg時,其降血糖作用最為顯著。這充分表明了,對GLP-1序列中的第8位氨基酸A突變為G時,其活性不受影響,依然能夠發揮降血糖作用。
而胰島素分泌試驗也表明,隨著濃度的增加,其胰島素分泌量也增加(圖9B)。表明,Z1Z2/GLTe-G復合物能有效刺激胰島細胞分泌胰島素,從而發揮降血糖作用。
2.體內穩定性檢測
將Z1Z2/GLTe-G復合物經靜脈注射入SD大鼠體內后,每隔一定時間尾部取血,用GLP-1(Total)Elisa Kit(Millipore)對血漿中Z1Z2/GLTe-G進行檢測(圖10)。發現其下降速度較慢,經計算其半衰期約為4h,是突變前(67min)的4倍,而是原GLP-1(1-2min)的約200倍,極大的延長了其半衰期,為更好的應用于臨床奠定了基礎。
實施例三多肽復合物作為SST的載體
生長抑素(Somatostatin,SST)是一種環狀多肽,廣泛分布于中樞神經系統和外周神經系統以及胃腸道中,能顯著抑制生長激素的分泌和釋放,也能抑制胰腺激素、胃泌素分泌等。SST能調控胰島素和胰高血糖素的分泌。SST有兩種活性形式,SST14和SST28。SST與屬于GPCRs的SST受體結合,從而激活下游通路。主要用于治療肢端肥大癥,肝硬化門脈高壓導致的食管靜脈出血,急性胰腺炎及其并發癥和胰膽腸瘺等。但是由于其在體內的半衰期只有2-3min(Afargan M et al,Endocrinology,2001,142(1):477-486),嚴重限制了其在臨床上的應用。
實驗部分:
1.基因融合
采用全基因合成的方式分別獲得實施例1中pET-Z1Z2和pET-TeN載體(也可以直接使用實施例1中的這兩個載體或其表達產物),以及具有將SST基因插入到TeN突變體Loop(LT1)中的基因序列的載體pET-24d-SSTe(原pET24d載體之外的序列部分如下SEQ ID NO:11)。
其中,箭頭所指為TEV酶酶切位點,雙下劃線為6xHis標簽,單下劃線為SST氨基酸序列,曲線部分為linker。
2.表達、純化、除內毒素
采用同實施例1的方法步驟進行,分別獲得純度較高且內毒素的含量小于2EU/ml的Z1Z2/TeN復合物和Z1Z2/SSTe復合物。
3.活性檢測
將250g左右的SD大鼠禁食12-16h后,隨機分為2組,一組為實驗組,按體重腹腔注射200nmol/kg的Z1Z2/SSTe復合物;另一組注射等體積的PBS。在0,30,60,120min時分別從尾部取血,加入到含有EDTA-Na2的離心管中,4000rpm離心10min,取上清。上清中生長激素的濃度用Growth hormone ELISA kit(Millipore)試劑盒檢測。
4.半衰期檢測
將Z1Z2/SSTe 0.28mg通過尾靜脈注射入大鼠體內,分別在0,0.5,1,2,4,8,24h,從尾靜脈取血,加入到含有EDTA-Na2的離心管中,4000rpm離心10min,取上清。上清中Z1Z2/SSTe中的SST用Somatostatin(SST)EIA kit(Phoenix pharmaceuticals)進行檢測。
結果與分析部分:
1.Z1Z2/SSTe表達純化
從圖11可以看出,經過一系列的純化后,Z1Z2/SSTe在SDS-PAGE圖上表現為兩條非常純的條帶;而在Native-PAGE圖上則表現為一條非常純的條帶,為Z1Z2/SSTe復合物。這表明,我們獲得了純度較高的用于后續試驗的Z1Z2/SSTe復合物。
Z1Z2/SSTe復合物的內毒素經內毒素去除樹脂處理后含量控制在2EU/ml的范圍內,可以用于后續實驗。
2.活性檢測
SST在體內能抑制生長激素的分泌,因此可以通過測定大鼠體內生長激素的含量來驗證Z1Z2/SSTe復合物的活性。Z1Z2/SSTe復合物注射入大鼠體內后,顯著抑制了大鼠生長激素的水平。SST及Z1Z2/SSTe注射入大鼠體內后,初期均能明顯抑制體內生長激素的分泌,但是SST的半衰期(2-3min)較短,隨著SST的降解,其抑制作用逐漸減弱,最后在2h時恢復到對照水平;而Z1Z2/SSTe能在120min內明顯抑制生長激素的水平,說明其具有更長的半衰期(圖12)。表明其具有與原SST類似的生物學活性,且能更長時間地發揮對生長激素的抑制作用。
3.半衰期檢測
Z1Z2/SSTe復合物經靜脈注射后,血漿中SST的含量如圖13所示,其計算的半衰期為6.62±0.43h,比原SST(2-3min)延長了大約200倍。這表明Z1Z2/SSTe復合物在不影響SST與其受體結合的情況下,極大延長了其半衰期,使其能更長時間發揮藥效。
- 還沒有人留言評論。精彩留言會獲得點贊!