一種具有多藥協同作用的抗腫瘤高分子鍵合藥及其制備方法與流程
本發明涉及一種抗腫瘤藥物,特別涉及一種具有多藥協同作用的抗腫瘤高分子鍵合藥及其制備方法;屬于生物醫藥高分子材料領域。
背景技術:
腫瘤是一個全球性的公共健康問題。近三十年來,世界癌癥發病率以每年3-5%的速度增加,二十一世紀,腫瘤已成為導致人類死亡的第一誘因。化療作為腫瘤三大治療手段之一,是目前腫瘤治療的重要手段。盡管化療在治療腫瘤方面已經取得了長足的進步,然而在臨床應用中仍存在一些障礙。如傳統的腫瘤化療采取的是單一化療藥物,其主要缺陷是單一藥物的治療往往會激活和強化腫瘤細胞其他方面的存活路徑,從而導致耐藥性的出現并最終導致化療失敗。同時為了達到治療效果,需要更高的藥物劑量,導致難以接受的藥物毒副作用。為了解決這一難題,近年來,利用多個藥物的協同作用使用聯合給藥的方法成為抗腫瘤藥物藥劑學領域的一個研究熱點。
目前,臨床使用的抗腫瘤藥物以紫杉醇、阿霉素、喜樹堿、順鉑為主。這類抗腫瘤藥物因其抗癌機理的不同而作用于腫瘤細胞的不同周期。如紫杉醇作用于微管/微管蛋白系統,可影響微管蛋白的裝配和解聚,從而導致微管束的排列異常,形成星狀體,使紡錘體失去正常功能,導致細胞死亡,作用于細胞周期的G2和M期;阿霉素通過嵌入癌細胞的DNA堿基片段中,阻礙DNA的轉錄和復制,從而抑制腫瘤細胞生長,屬周期非特異性藥物;喜樹堿能穩定拓撲異構酶I和DNA的共價化合物,形成了三元可解離復合物,通過復制沖突模型造成DNA損傷,最終導致細胞死亡,作用于細胞周期的S期。聯合用藥是基于不同的化療藥物對應于腫瘤細胞的不同信號通路,在腫瘤細胞的不同生長周期產生抑制作用,從而實現治療目的。
然而小分子藥物的聯合存在不同藥物的代謝性質、組織和器官分布、穿透各 級膜的能力的不同的問題,很難控制到達最終靶點的多種藥物仍保持注射時的最初濃度。其次,小分子藥物的聯合,往往會帶來更大的副作用和毒性。因此,使用聚合物前藥或納米藥物載體成為聯合用藥的最佳策略。眾所周知,納米藥物載體具有可通過合適的尺寸和結構設計來改變藥物的藥代動力學和體內分布以及通過EPR效應實現被動靶向的優點。作為載體給藥系統的一員,從兩親性嵌段共聚物發展而來的聚合物膠束深受青睞。當投入到水性介質中時,在體系自由能降低的驅動下,膠束的疏水段自發聚集在一起形成微粒的內核。它既可以作為很多難溶性藥物的微儲庫,又能避免藥物在生物體內環境中失活。自組裝形成的載藥膠束是熱力學、動力學穩定的體系,具有穩定、長效、安全等許多優良的性質,使得聚合物膠束成為難溶性藥物理想的輸送系統,且在藥物釋放、基因載體和診斷制劑等很多方面得到廣泛地應用。
聚合物修飾劑的選擇是藥物分子修飾的關鍵,現有研究表明脂肪族聚酯如聚乳酸(PLA),聚乳酸-羥基乙酸共聚物(PLGA)和聚ε-己內酯(PCL)屬于生物可降解聚合物。它們的分子量在相當寬的范圍可以隨意控制,所以在聚合物藥物載體的研究中得到了廣泛的應用。特別是在與水溶性聚合物形成嵌段共聚物后,不僅具有生物可降解性,而且大大地改善了材料與人體的生物相容性,作為藥物載體材料時,延長了藥物在體內的循環時間,提高了藥效,降低了免疫響應性,已經成為生物醫藥高分子材料領域的研究熱點。
技術實現要素:
針對傳統的腫瘤化療采取的是單一化療藥,或一些聯合給藥的藥物載體大多都是不可降解的高分子聚合物,且其鍵合方式都是物理包裹,導致人體對藥物的生物利用度低,抗癌活性差等缺陷;本發明的目的是在于提供一種以可生物降解高分子為載體,同時負載多種抗腫瘤藥物構成的高分子鍵合藥,該高分子鍵合藥通過多藥協同作用提高抗癌活性,同時降低化療的毒副作用,且該高分子鍵合藥生物相容性好,載藥量高且可控,可以應用于臨床研究。
本發明的另一個目的是在于提供一種經濟、高效和無毒的制備上述可生物降解的具有多藥協同作用的抗腫瘤藥高分子鍵合藥的方法。
為了實現上述技術目的,本發明提供了一種具有多藥協同作用的抗腫瘤高分 子鍵合藥,具有式1結構:
其中,
A為含有羥基的抗腫瘤藥物基團;
B為含有氨基的抗腫瘤藥物基團;
n為45~454,x為1~20,y為1~20;
R1和R2獨立地選自C1~C4的亞烷基。
優選的方案,A為紫杉醇基團、多西紫杉醇基團、喜樹堿基團、絲裂霉素C基團中的至少一種。
優選的方案,B為阿霉素基團、表阿霉素基團、吡喃阿霉素基團中的至少一種。
本發明還提供了一種制備所述的具有多藥協同作用的抗腫瘤高分子鍵合藥的方法,該方法包括以下步驟:
1)以聚乙二醇單甲醚引發含降冰片烯基團的丙交酯進行開環聚合,聚合產物再引發α溴代己內酯進行開環聚合,得到聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物;
2)所述聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與含巰基的醇類化合物進行巰基-溴點擊反應,得到含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物;所述含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與對硝基苯基氯甲酸酯進行縮合反應,得到含對硝基苯基甲酸 酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物;所述含對硝基苯基甲酸酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與含巰基的酸類化合物進行巰基-烯光點擊反應,得到式2載體聚合物;
3)含羥基的抗腫瘤藥物和含氨基的抗腫瘤藥物與式2載體聚合物分別進行酯化反應和酰胺化反應,即得;
其中,
n為45~454;
x為1~20;
y為1~20;
R1和R2各自獨立地選自C1~C4的亞烷基。
優選的方案,含有氨基的抗腫瘤藥物為阿霉素、表阿霉素、吡喃阿霉素中的至少一種。
優選的方案,含有羥基的抗腫瘤藥物為紫杉醇、多西紫杉醇、喜樹堿、絲裂霉素C中的至少一種。
本發明的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯聚合物的制備方法:
將丙交酯和N-溴代丁二酰亞胺(NBS),以四氯化碳或苯為溶液,以過氧化二苯甲酰(BPO)做催化劑,在60~90℃下發生取代反應,得到溴代丙交酯;得到的溴代丙交酯以二氯甲烷溶劑中,在三乙胺的作用下,0~5℃下發生消去反應得到 雙鍵丙交酯;得到的雙鍵丙交酯和新蒸環戊二烯在四氯化碳或苯溶液中,氬氣保護,60~90℃下,通過Diels-Alder反應后,得到含降冰片烯側基的丙交酯;
將環己酮和N-溴代丁二酰亞胺(NBS)以無水乙醚為溶劑,醋酸銨(NH4AcO)做催化劑,室溫下反應得到溴代環己酮;溴代環己酮通過Baeyer-Villiger氧化反應與間氯過氧苯甲酸(m-CPBA)生成了α位溴代己內酯;
以聚乙二醇單甲醚為大分子引發劑,TBD或DBU為催化劑,二氯甲烷為溶劑,-20~40℃下引發含降冰片烯側基的丙交酯進行開環聚合得到聚乙二醇單甲醚-b-聚丙交酯聚合物,再以聚乙二醇單甲醚-b-聚丙交酯聚合物為大分子引發劑,同樣的聚合條件將α溴代己內酯引發聚合,得到聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯聚合物。
本發明的聚合物官能化的制備方法:將聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯聚合物采用巰基-溴點擊化學反應和巰基-烯光點擊化學反應,反應點擊效率高,無副產物。
本發明的含羥基的抗腫瘤藥物與高分子載體進行DIC縮合反應,其副產物易除去,避免了傳統DCC縮合后劇毒物質DCU的產生。含氨基的抗腫瘤藥物與對硝基苯基甲酸酯活化的高分子載體鍵合,其鍵合效率高,生成的酰胺鍵穩定。
本發明的具有多藥協同作用的抗腫瘤高分子鍵合藥的合成路線如下:
以抗腫瘤藥物紫杉醇(paclitaxel)和阿霉素(doxorubicin)為例:
相對現有技術,本發明的技術方案帶來的有益效果:
1)與現有的高分子抗腫瘤高分子鍵合藥相比,本發明的抗腫瘤高分子鍵合藥最大的優勢在于通過將抗腫瘤藥物阿霉素和紫杉醇等同時鍵合到高分子載體上,不同的抗腫瘤藥物對應于腫瘤細胞的不同信號通路,在腫瘤細胞的不同生長周期產生抑制作用,提高治療效果。如阿霉素通過嵌入癌細胞的DNA堿基片段 中,阻礙DNA的轉錄和復制,從而抑制腫瘤細胞生長;而紫杉醇作用于微管/微管蛋白系統,可影響微管蛋白的裝配和解聚,從而導致微管束的排列異常,形成星狀體,使紡錘體失去正常功能,導致細胞死亡。
2)本發明的高分子抗腫瘤高分子鍵合藥另一優勢在于可降低化療藥物的毒副作用,因為藥物聯合作用導致每種藥物的給藥劑量減少,同時抗癌活性不受影響,提高了藥物的利用度,可以降低化療成本。
3)本發明的高分子抗腫瘤高分子鍵合藥在端基引入分子量較小的聚乙二醇單甲醚,聚乙二醇單甲醚不但具有較好的生物相容性及生物可降解性,并且在水相中易于自組裝形成納米膠束粒子,延長了藥物在體內的循環時間,提高了藥效,降低了免疫響應性。
4)本發明的高分子抗腫瘤高分子鍵合藥的制備方法操作簡單、反應條件溫和、副反應少、產率高和安全無毒,且易于調控高分子鍵合藥的載藥量,滿足工業生產要求。
附圖說明
【圖1】為聚乙二醇單甲醚-b-聚丙交酯兩嵌段共聚物的核磁氫譜圖;
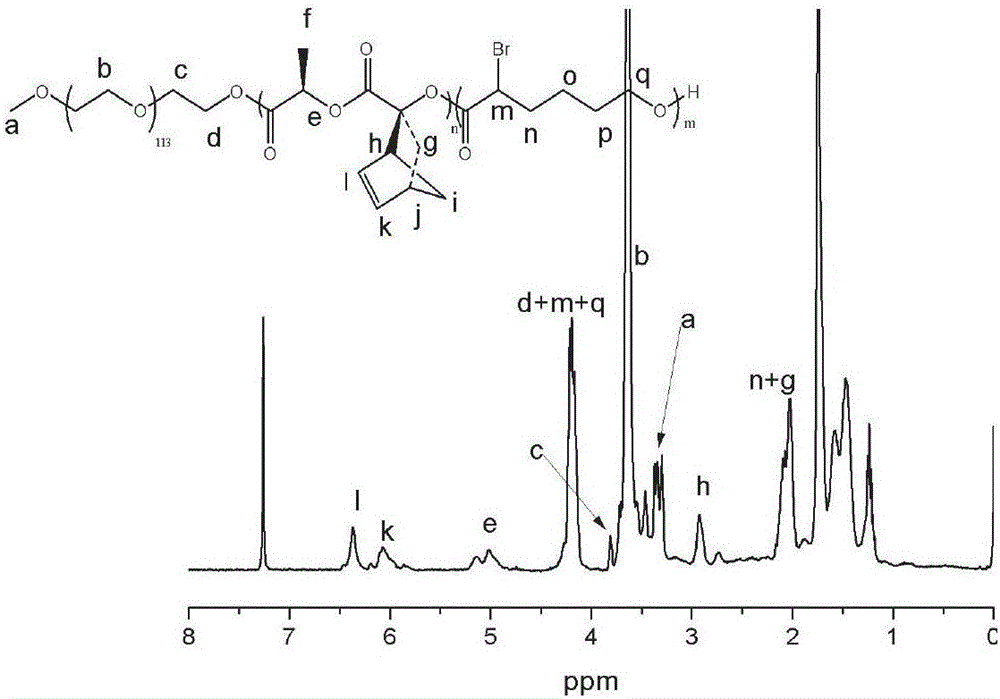
【圖2】為聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的核磁氫譜圖;【圖3】為實施例1的步驟1中各聚合物的分子量分布圖:A為聚乙二醇單甲醚的分子量分布圖,B為聚乙二醇單甲醚-b-聚丙交酯共聚物的分子量分布圖,C為聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的分子量分布圖;
【圖4】為聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與巰基乙醇反生巰基-溴點擊反應得到的含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物核磁氫譜圖;
【圖5】為含對硝基苯基甲酸酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的核磁氫譜圖;
【圖6】為含對硝基苯基甲酸酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與巰基丙酸反生巰基-烯光點擊反應得到的載體聚合物的核磁氫譜圖;
【圖7】為含紫杉醇的抗腫瘤高分子鍵合藥的核磁氫譜圖;
【圖8】為含紫杉醇和阿霉素高分子鍵合藥的核磁氫譜圖;
【圖9】為實施例1中步驟2~4中各聚合物的分子量分布圖:A為聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與巰基乙醇反生巰基-溴點擊反應得到的含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的分子量分布圖,B為含對硝基苯基甲酸酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的分子量分布圖,C為含對硝基苯基甲酸酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物與巰基丙酸反生巰基-烯光點擊反應得到的含羧基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物分子量分布圖。
具體實施方式
以下實施例旨在是對本發明內容進一步說明,而不是限制本發明權利要求的保護范圍。
實施例1
1、聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的制備:
首先,在己真空烤過3次的安培瓶中,于氮氣保護下加入0.80g的降冰片烯側基的丙交酯和1.00g聚乙二醇單甲醚(mPEG),之后加入1mL的精制的DCM使其完全溶解,注射由精制DCM配制好的含引發劑TBD溶液1mL,常溫反應48h后,用無水乙醚沉降3次,離心后真空干燥,得到聚乙二醇單甲醚-b-聚丙交酯兩嵌段聚合物,結構表征如圖1所示,分子量分布為圖3。然后,再以其為大分子引發劑,同樣聚合條件下引發α溴代己內酯聚合,得到聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物,其結構表征如圖2,分子量分布見圖3,說明該聚合物已成功合成。
2、含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的制備:
稱取上述聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物0.5g(0.06mmol),2-巰基乙醇0.054g(0.73mmol),溶于10mL的乙腈,加入0.05mL三乙胺做催化劑,在氬氣保護下反應2h。反應結束后用水透析24h,除去季銨鹽,再用無水乙醚沉降,離心后真空干燥,得到純的含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物。其結構表征見核磁氫譜圖(圖4),分子量分 布見圖9A,說明該聚合物已成功合成。
3、含對硝基苯基甲酸酯側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯共聚物的制備:
稱取含羥基側基的聚乙二醇單甲醚-b-聚丙交酯-b-聚α溴代己內酯聚合物0.4g(0.049mmol),溶于10mL二氯甲烷,在氬氣保護下緩慢滴加0.21g對硝基苯基氯甲酸酯和0.05mL三乙胺,0℃反應24小時,反應結束后用無水乙醚沉降3次,離心后真空干燥。其結構表征見核磁氫譜圖6,分子量分布見圖9B。
4、載體聚合物的制備:
在氮氣的保護下,將0.3g(0.45mmol)上述聚合物溶解在10mL精制的四氫呋喃(THF)中,加入0.030g(0.029mmol)3-巰基丙酸,3mg安息香二甲醚(DMPA)光引發劑,在紫外燈光照下反應0.5h。反應結束后,用無水乙醚沉降3次,離心后真空干燥,得到載體聚合物。其結構表征見核磁氫譜圖6,分子量分布見圖9C,說明該聚合物已成功合成。
5、含紫杉醇的高分子鍵合藥的制備
稱取載體聚合物0.1g(0.008mmol),紫杉醇0.029g(0.03mmol),N,N-二異丙基碳二亞胺(DIC)0.004g(0.03mmol)。將紫杉醇溶于10mL三氯甲烷,加入催化量的DMAP,在氮氣保護下緩慢滴加溶于2mL三氯甲烷的聚合物和DIC,冰浴攪拌1h后,在45℃下反應24h。反應結束后用甲醇透析24h,除掉脲鹽,然后用無水乙醚沉降3次,離心,真空干燥。其結構表征見核磁氫譜圖7。
6、含紫杉醇和阿霉素的高分子鍵合藥的制備:
稱取上述紫杉醇前藥0.5g,鹽酸鹽阿霉素0.025g,將鹽酸鹽阿霉素溶于干燥的DMF,加入0.01mL三乙胺,抽真空充氮氣三次,反應2小時脫掉鹽酸鹽,再加入紫杉醇前藥,避光,室溫反應48h。反應結束后,用DMF透析48h,乙腈透析掉DMF,無水乙醚沉降,離心,真空干燥。其結構表征見核磁氫譜圖8。
- 還沒有人留言評論。精彩留言會獲得點贊!