一種新型的CCR2拮抗劑及其用途的制作方法
本申請涉及一種新型的ccr2拮抗劑及其用途。
背景技術:
如cn103328479a中所述,趨化因子受體ccr2見于單核細胞、巨噬細胞、b細胞、活化的t細胞、樹突細胞、內皮細胞和腫瘤細胞的表面上。它是多種趨化因子配體(包括mcp-1、mcp-2、mcp-3和mcp-4)的受體。其中,mcp-1(單核細胞趨化蛋白-1,也稱為ccl2)似乎僅與ccr2相互作用,而不與迄今為止鑒定的任何其他趨化因子受體相互作用。mcp-1由心肌細胞、血管內皮細胞、成纖維細胞、軟骨細胞、平滑肌細胞、系膜細胞、肺泡細胞、t淋巴細胞、巨噬細胞等表達。在被其mcp-1活化后,ccr2受體信號級聯放大涉及磷脂酶、蛋白激酶和脂質激酶的活化。
ccr2介導的單核細胞募集是導致發生動脈粥樣硬化的最早期步驟之一。在動脈粥樣硬化的實驗模型中,動脈斑塊形成依賴于ccr2和mcp-1的完整性。
慢性炎性疾病的動物模型研究已證明,通過拮抗劑抑制mcp-1與ccr2之間的結合抑制了炎性應答。已表明抑制發生關節炎、哮喘和葡萄膜炎的mcp-1拮抗劑抑制單核細胞遷移。mcp-1和ccr2二者敲除小鼠已證明,單核細胞向炎性病變中的浸潤顯著降低。單核細胞的ccr2介導性遷移被認為是人多發性硬化(ms)的致病原因,因為在ms患者的腦脊液中觀察到了ccr2和mcp-1表達。在人ms的小鼠模型中,ccr2或mcp-1缺陷預防eae的發生。在類風濕性關節炎和克羅恩病中用tnf-α拮抗劑治療期間觀察到的改善還與mcp-1表達和浸潤性巨噬細胞數目的降低相關。另外,最近有人提出了ccr2影響肥胖和相關脂肪組織炎癥和全身胰島素抗性的發生,并且在一旦確立肥胖及其代謝結果后在維持脂肪組織巨噬細胞和胰島素抗性中發揮作用。此外,ccr2信號傳導可在神經性疼痛中發揮致病作用。還表明缺乏ccr2使小鼠疼痛模型中的炎性和神經性疼痛降低,表明神經組織中巨噬細胞和小膠質細胞的募集和活化在疼痛狀態中發揮重要作用。
已將mcp-1與ccr2之間的相互作用與炎性疾病的病理學聯系起來,所述疾病例如銀屑病、葡萄膜炎、動脈粥樣硬化、類風濕性關節炎、多發性硬化、克羅恩病、炎性腸病、腎炎、器官同種異體移植排斥、纖維化肺、腎功能不全、腎纖維化、糖尿病和糖尿病并發癥、糖尿病性腎病、糖尿病性視網膜病、糖尿病性視網膜炎、糖尿病性微血管病、肥胖、神經性疼痛(包括與糖尿病相關的神經性疼痛)、結核病、結節病、侵襲性葡萄球菌感染、白內障手術后炎癥、變應性鼻炎、變應性結膜炎、慢性蕁麻疹、慢性阻塞性肺病(copd)、變應性哮喘、hiv相關癡呆癥、牙周病、牙周炎、牙齦炎、齒齦病、舒張性心肌病、心肌梗死、心肌炎、慢性心力衰竭、血管狹窄、再狹窄、再灌注障礙、腎小球腎炎、實體瘤和癌癥、慢性淋巴細胞性白血病、慢性髓細胞性白血病、多發性骨髓瘤、惡性骨髓瘤、霍奇金病、以及膀胱癌、乳腺癌、子宮頸癌、結腸癌、直腸癌、肺癌、前列腺癌和胃癌(參見,例如rollins,mol.med.today,2:198-204(1996);dawson等,expertopin.ther.targets,7(1):35-48,(2003)),connor等,gut,153:1287-1294;ali-osmanjr等,j.surg.res.,144:350-351(2008);cid等,rheumatology,45(11):1356-1363(2006);wada等,inflammationandregeneration,23(5):567-572(2004)。
ccr2和mcp-1與多種癌癥的生長、確立和轉移擴散密切相關(yoshie等,2001)。一般而言,認為ccr2介導的巨噬細胞和免疫抑制性骨髓細胞對腫瘤和轉移細胞的吸引是所涉及的主要機制,但是多種骨髓祖細胞的活動化也可發揮作用。特別有趣的是,在許多腫瘤類型中,mcp-1水平與侵襲、巨噬細胞含量和血管發生相關聯。血漿ccl2水平在癌癥患者中傾向于升高并且與乳腺癌(dwyer等,2007)、卵巢癌(hefler等,1999)和肺癌(cai等,2009)患者的腫瘤階段相關。
ccr2受體和mcp-1/ccl2的多態性與人的癌癥發生率顯著相關,包括膀胱癌和子宮頸癌(zafiropoulos等,2004;coelho等,2005;narter等,2010)。其中與mcp-1和ccr2相關的癌癥包括黑素瘤(graves等,1992;koga等2008;zheng等,1999,)、卵巢癌(negus等,1995)、乳腺癌(saji等,2001;soria等,2008;soria和benbaruch2008;mestdagt等2004;chavey等,2007;valkovic等,1998;ueno等,2000;valkovic等,2005;salcedo等,2000)、食管癌(ohta等,2002;koide等,2004)、胃癌(ohta等,2003;kuroda等,2005;futagami等,2008)、腎細胞癌(lukesova等,2008)、肺癌(cai等,2009;wong等,2008;niiya等,2003)、結腸癌(bailey等,2007)、甲狀腺癌(tanaka等,2009)、白血病(mazur等,2007)、多發性骨髓瘤(arendt等,2002;johrer等,2004;vandebroeke等,2003;pellegrino等,2005)和前列腺癌(lu等,2007a)。
mcp-1促進前破骨細胞融合,導致形成破骨細胞(lu等,2007b),并且還參與促進cd11b+細胞分化為破骨細胞(mizutani等,2009)。數種癌主要轉移至骨,包括肺癌、乳腺癌、腎癌、甲狀腺癌和多發性骨髓瘤(參見craig和loberg,2006)。多于90%的晚期前列腺癌患者表現出骨轉移的跡象(shah等,2004)。
mcp-1在骨定向轉移的發生中發揮核心作用。lu和kang(2009)表明,使用提高mcp-1表達的人乳腺腫瘤系促進了肺轉移和骨轉移以及繼發性腫瘤的后續生長。
肝是結直腸癌轉移的主要部位。然而,肝切除術很少有療效,有60~70%的病例發生復發。mcp-1可在肝轉移中高度表達,并且高的水平與不良預后相關,mcp-1表達隨癌癥分期而明顯升高(bailey等,2007;yoshimode等,2009)。
與肝腫瘤相關的成纖維細胞和正常成纖維細胞在tnfα的影響下均表達mcp-1(muller等,2007,2010),表明與腫瘤相關的成纖維細胞來自炎性條件下的正常肝基質。
因此,大量證據證明,mcp-1和ccr2參與癌癥的生長和發展,特別是癌癥相關細胞(如巨噬細胞)的募集。因此,ccr2拮抗劑應可用于治療癌癥,尤其是用于限制癌細胞轉移擴散,并且降低巨噬細胞和骨髓細胞向原發性腫瘤的募集,從而降低腫瘤生長和血管形成。特別是,ccr2拮抗劑應可用于抑制轉移性腫瘤細胞從原發性腫瘤部位的擴散。
技術實現要素:
本發明第一方面提供下式i所示的化合物或其藥學上可接受的鹽在制備藥物中的用途,尤其是在制備用于治療或預防ccr2介導的疾病的藥物中的用途:
式中,
r1、r2、r3和r4各自獨立為oh或-oc(o)r5,其中,r5選自c1-c4烷基;
a、b、c和d各自獨立為0、1、2或3的整數。
在一個或多個實施方案中,r1、r2、r3和r4各自獨立為oh。
在一個或多個實施例中,a為2,b、c和d為1。
在一個或多個實施方案中,b、c和d為1,r2、r3和r4各自獨立地位于對位或間位上。
在一個或多個實施方案中,式i化合物具有下式ii所示的結構:
式中,a、b、c、d、r1、r2、r3和r4各自如前文所述。
在一個或多個實施方案中,式ii化合物中,a為2,b、c和d為1,兩個r1分別位于5和7位,r2、r3和r4各自獨立地位于對位上。
在一個或多個實施方案中,式i化合物為下式化合物:
在一個或多個實施方案中,所述ccr2介導的疾病包括但不限于疼痛、炎性疾病、癌癥、癌細胞轉移擴散等。
在一個或多個實施方案中,所述ccr2介導的疾病包括但不限于銀屑病、葡萄膜炎、動脈粥樣硬化、類風濕性關節炎、多發性硬化、克羅恩病、炎性腸病、腎炎、器官同種異體移植排斥、纖維化肺、腎功能不全、腎纖維化、糖尿病和糖尿病并發癥、糖尿病性腎病、糖尿病性視網膜病、糖尿病性視網膜炎、糖尿病性微血管病、肥胖、神經性疼痛(包括與糖尿病相關的神經性疼痛)、結核病、結節病、侵襲性葡萄球菌感染、白內障手術后炎癥、變應性鼻炎、變應性結膜炎、慢性蕁麻疹、慢性阻塞性肺病、變應性哮喘、hiv相關癡呆癥、牙周病(牙周炎、牙齦炎、齒齦病)、舒張性心肌病、心肌梗死、心肌炎、慢性心力衰竭、血管狹窄、再狹窄、再灌注障礙、腎小球腎炎、實體瘤和癌癥、慢性淋巴細胞性白血病、慢性髓細胞性白血病、多發性骨髓瘤、惡性骨髓瘤和霍奇金病。
在一個或多個實施方案中,所述ccr2介導的疾病包括但不限于膀胱癌、子宮頸癌、黑素瘤、卵巢癌、乳腺癌、食管癌、胃癌、腎細胞癌、肺癌、結腸癌、直腸癌、甲狀腺癌、白血病、多發性骨髓瘤、肝癌和前列腺癌。
在一個或多個實施方案中,所述ccr2介導的疾病包括轉移性腫瘤細胞從原發性腫瘤部位的擴散,例如,肺癌、乳腺癌、腎癌、甲狀腺癌和多發性骨髓瘤的癌細胞向骨的轉移,乳腺癌細胞向肺的轉移,結腸癌、直腸癌細胞向肝的轉移。
在一個或多個實施方案中,所述藥物還包括一種或多種已知的抗癌藥物。
在一個或多個實施方案中,所述已知的抗癌藥物選自:白消安、馬法蘭、苯丁酸氮芥、環磷酰胺、異環磷酰胺、替莫唑胺、苯達莫司汀、順鉑、絲裂霉素c、博萊霉素、卡鉑、喜樹堿、伊立替康、托泊替康、阿霉素、表阿霉素、阿克拉霉素、米托蒽醌、甲基羥基玫瑰樹堿、5-氮雜胞苷、吉西他濱、5-氟尿嘧啶、5-氟-2'-去氧尿苷、氟達拉濱、奈拉濱、長春新堿、長春瑞濱、紫杉醇、伊沙匹隆、卡巴他賽、多西他賽、帕尼單抗、阿瓦斯丁、赫賽汀、美羅華、伊馬替尼、吉非替尼、埃羅替尼、拉帕替尼、甲苯磺酸索拉非尼、舒尼替尼、尼羅替尼、達沙替尼、帕唑帕尼、依維莫司、他莫昔芬、來曲唑、氟維司群和米托胍腙。
在一個或多個實施方案中,所述已知的抗癌藥物為甲苯磺酸索拉非尼。
本發明還提供式i所示的化合物或其藥學上可接受的鹽在制備用于在需要的對象中調節腫瘤微環境、降低巨噬細胞和骨髓細胞向原發性腫瘤的募集、抑制腫瘤生長、抑制血管形成、和/或增加cd8t細胞的藥物或試劑中的用途。
在一個或多個實施方案中,所述用途包括制備用于下述任意一種或多種目的藥物或試劑的用途:
抑制ccr2介導的細胞遷移,包括但不限于抑制thp-1的遷移能力,或抑制巨噬細胞的招募;
抑制ccl2-ccr2引起的鈣流;
解除巨噬細胞對cd8t細胞的抑制作用;和
使巨噬細胞從免疫抑制型(m2)向免疫促進型(m1)偏移。
本發明還提供式i所示的化合物或其藥學上可接受的鹽在制備用于抑制ccr2或ccl2-ccr2通路的藥物或試劑中的用途。
附圖說明
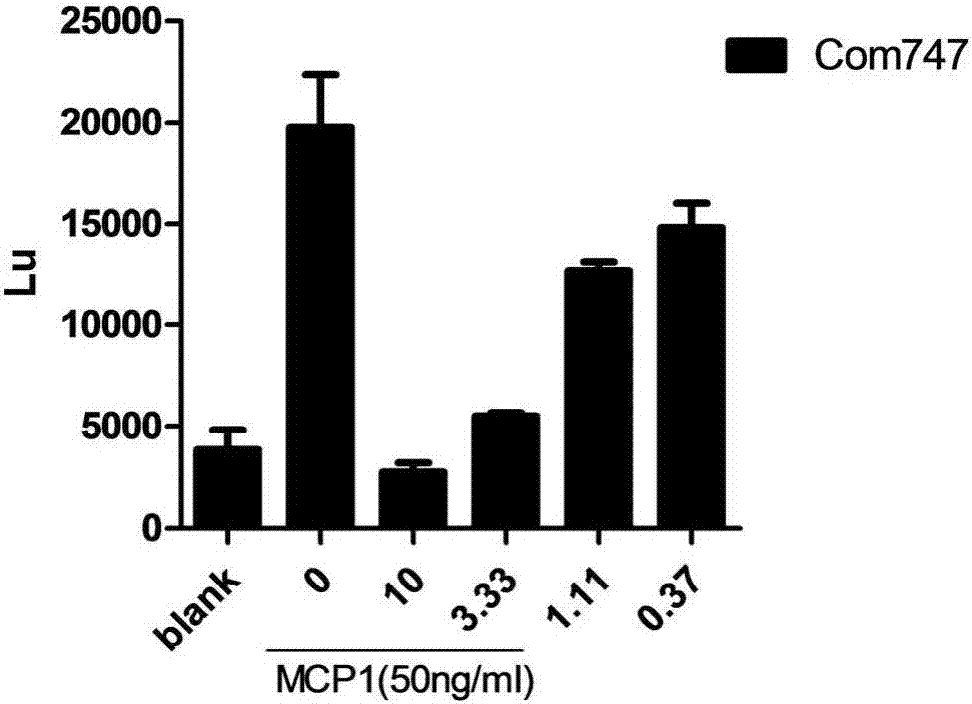
圖1:不同濃度747藥物對于ccl2-ccr2介導的thp-1遷移能力的影響。
圖2:10μm747與其他ccr2拮抗劑對thp1-gcamp6鈣流影響。
圖3:不同濃度747對293ft-gcamp6-ccr2鈣流影響。
圖4:不同濃度747對人與鼠的i125mcp1-ccr2的抑制能力。
圖5:不同濃度747對人與鼠的i125mip1a-ccr5的抑制能力。
圖6:747對tg腹膜炎誘導的腹腔巨噬細胞數量影響。
圖7:747對于多種肝癌細胞系體外增殖影響(48小時)。
圖8:747與sorafenib對于hepa1-6小鼠肝原位腫瘤治療活體成像追蹤。
圖9:747與sorafenib治療后hepa1-6肝原位腫瘤生長結果。
圖10:747與sorafenib治療hepa1-6肝原位腫瘤小鼠體重變化追蹤。
圖11:747對hepa1-6肝原位腫瘤微環境中cd8t細胞與cd4t細胞比例影響。
圖12:747、巨噬細胞敲除、cd8t細胞敲除對hepa1-6小鼠肝癌皮下腫瘤治療結果。
圖13:747、巨噬細胞敲除、cd8t細胞敲除對hepa1-6小鼠肝癌皮下腫瘤生長變化。
圖14:747對hepa1-6小鼠肝癌皮下腫瘤微環境cd4t細胞與cd8t細胞比例影響。
圖15:747對hepa1-6小鼠肝癌皮下腫瘤微環境巨噬細胞比例影響。
圖16:747、sorafenib各組對lpc小鼠皮下肝癌治療結果。
圖17:747、sorafenib各組對lpc小鼠肝癌皮下腫瘤生長變化追蹤。
圖18:747、sorafenib治療lpc小鼠肝癌皮下腫瘤后腫瘤重量。
圖19:747對lpc小鼠肝癌皮下腫瘤微環境cd8t細胞與cd4t細胞比例影響。
圖20:747、sorafenib對lpc小鼠肝癌皮下腫瘤微環境巨噬細胞的細胞比例。
圖21:lpc小鼠皮下腫瘤微環境的巨噬細胞中ccr2陽性細胞的各組比例。
圖22:lpc小鼠皮下腫瘤微環境的巨噬細胞中m2-免疫抑制性巨噬細胞的各組比例。
圖23:化合物747的hplc-uv檢測(a)和ms(+/-)(b)。該化合物該化合物在負模式下離子化好;正模式下離子化差。
具體實施方案
本文提供一類新的ccr2拮抗劑,其結構如前文所述的式i所示。優選的,式i化合物具有式ii所示的結構。
在優選的式i和ii化合物中,r1、r2、r3和r4各自獨立為oh;和/或,a為2,b、c和d為1;和/或,r2、r3和r4各自獨立地位于對位或間位上。
在某些優選的式i和ii化合物中,b、c和d為1,r2、r3和r4各自獨立為oh,且位于對位上;a為2。
在其它實施方案中,式i和ii化合物中,a為2,b、c和d為1,兩個r1分別位于5和7位,r2、r3和r4各自獨立地位于對位上。
更優選的,式i化合物為本文所述的編號為747的化合物(山奈酚3-(2,4-二-e-p-香豆酰基鼠李糖苷):
一些化合物可能作為立體異構體,包括旋光異構體存在。本申請包括所有立體異構體和這樣的立體異構體的外消旋混合物,以及可以根據本領域技術人員眾所周知的方法分離出來的單獨的對映體。
本文中,化合物747可從長苞冷杉中提取獲得。例如,在一個具體的提取方法中,將長苞冷杉干燥的地上部分粉碎成粗粉,用乙醇回流提取數次后,合并提取液,減壓濃縮成稠浸膏。浸膏加水稀釋后分別以氯仿、乙酸乙酯和正丁醇依次萃取,收集各萃取部分,分別濃縮成浸膏。其中乙酸乙酯萃取部分的浸膏,以甲醇溶解過濾后進行硅膠柱色譜分離,可獲得化合物747。
色譜分離時,可使用氯仿-甲醇(例如2:1的氯仿:甲醇)洗脫,用薄層色譜檢查洗脫流分,得流分1到10,其中流分1經進一步硅膠色譜純化,得到黃色無定形粉末,該粉末即為化合物747。
獲得化合物747后,可將其制備成相應的衍生物,例如,可采用本領域周知的方法將其制備成其藥學上可接受的鹽或酯。
本文中,藥學上可接受的鹽的例子包括但不限于無機和有機酸鹽,例如鹽酸鹽、氫溴酸鹽、磷酸鹽、硫酸鹽、檸檬酸鹽、乳酸鹽、酒石酸鹽、馬來酸鹽、富馬酸鹽、扁桃酸鹽和草酸鹽;以及與堿例如鈉羥基、三(羥基甲基)胺基甲烷(tris,胺丁三醇)和n-甲基葡糖胺形成的無機和有機堿鹽。
本文中,藥學上可接受的酯包括依據本領域已知方法通過使式i化合物上的羥基與c1-4羧酸、c3-6二酸或其酸酐例如琥珀酸酐和富馬酸酐縮合而獲得的酯。
作為ccr2拮抗劑,本發明式i化合物或其藥學上可接受的鹽可用于治療或預防ccr2介導的各種疾病,包括但不限于疼痛、炎性疾病、癌癥和癌細胞轉移擴散等。
舉例而言,ccr2介導的疾病包括但不限于銀屑病、葡萄膜炎、動脈粥樣硬化、類風濕性關節炎、多發性硬化、克羅恩病、炎性腸病、腎炎、器官同種異體移植排斥、纖維化肺、腎功能不全、腎纖維化、糖尿病和糖尿病并發癥、糖尿病性腎病、糖尿病性視網膜病、糖尿病性視網膜炎、糖尿病性微血管病、肥胖、神經性疼痛(包括與糖尿病相關的神經性疼痛)、結核病、結節病、侵襲性葡萄球菌感染、白內障手術后炎癥、變應性鼻炎、變應性結膜炎、慢性蕁麻疹、慢性阻塞性肺病、變應性哮喘、hiv相關癡呆癥、牙周病(牙周炎、牙齦炎、齒齦病)、舒張性心肌病、心肌梗死、心肌炎、慢性心力衰竭、血管狹窄、再狹窄、再灌注障礙、腎小球腎炎、實體瘤和癌癥、慢性淋巴細胞性白血病、慢性髓細胞性白血病、多發性骨髓瘤、惡性骨髓瘤、癌細胞轉移擴散和霍奇金病。
就癌癥的治療和預防而言,本申請式i化合物通過ccr2這一免疫靶點調節了腫瘤微環境,減少了腫瘤巨噬細胞的浸潤,增加了cd8t細胞,從而達到治療腫瘤的效果。因此,本申請包括式i化合物或其藥學上可接受的鹽用于下述任意一種或多種目的的用途或方法(包括制備用于下述一種或多種目的的藥物組合物/試劑的用途):
調節腫瘤微環境;
減少腫瘤巨噬細胞浸潤;和
增加cd8t細胞。
更具體而言,本申請包括式i化合物或其藥學上可接受的鹽用于下述任意一種或多種目的的用途或方法(包括制備用于下述一種或多種目的的藥物組合物/試劑的用途):
抑制ccr2介導的細胞遷移,包括但不限于抑制thp-1的遷移能力,或抑制巨噬細胞的招募;
抑制ccl2-ccr2引起的鈣流;
解除巨噬細胞對cd8t細胞的抑制作用;和
使巨噬細胞從免疫抑制型(m2)向免疫促進型(m1)偏移。
本文中,ccr2介導的癌癥包括但不限于膀胱癌、子宮頸癌、黑素瘤、卵巢癌、乳腺癌、食管癌、胃癌、腎細胞癌、肺癌、結腸癌、直腸癌、甲狀腺癌、白血病、多發性骨髓瘤、肝癌和前列腺癌。
本文中,癌細胞轉移擴散包括但不限于轉移性腫瘤細胞從原發性腫瘤部位的擴散,例如,肺癌、乳腺癌、腎癌、甲狀腺癌和多發性骨髓瘤向骨的轉移,乳腺癌細胞向肺的轉移,結腸癌、直腸癌細胞向肝的轉移。
本申請式i化合物或其藥學上可接受的鹽也可用于降低巨噬細胞和骨髓細胞向原發性腫瘤的募集、抑制腫瘤生長和/或抑制血管形成,因此,本申請也包括式i化合物或其藥學上可接受的鹽在制備用于降低巨噬細胞和骨髓細胞向原發性腫瘤的募集、抑制腫瘤生長和/或抑制血管形成的藥物組合物或試劑中的用途。
本申請特別包括化合物747及其藥學上可接受的鹽在制備治療或預防ccr2介導的癌癥以及癌細胞轉移擴散的藥物中的用途,尤其是在制備治療或預防肝癌(如ccr2介導的肝癌)的藥物中的用途。
本申請還包括用于治療或預防上述疾病的本申請式i化合物,尤其是式ii化合物和化合物747。
因此,本申請也包括上述疾病的治療或預防方法,所述方法包括給予需要的對象治療或預防有效量的本申請式i化合物。尤其是,本申請包括肝癌(如ccr2介導的肝癌)的治療或預防方法,上述方法包括給予需要的對象治療或預防有效量的化合物747。
本文中,治療或預防有效量可由本領域技術人員根據本領域常規的方法容易地確定。例如,一般情況下,式i化合物或其藥學上可接受的鹽對哺乳動物每天口服給藥,藥量可按照約0.0025到50毫克/公斤體重。單位口服劑量可以包括約0.01到50毫克式i化合物。單位劑量可給予一次或多次,每天為一片或多片,每片含有約0.1到50毫克。
對于上述用途或方法而言,本申請式i化合物可以藥物組合物或試劑的方式給予或施用。
藥物組合物中通常含有本領域常規施用的藥學上可接受的載體。藥學上可接受的載體包括藥學上可接受的賦形劑和稀釋劑。載體可為惰性的或者其本身可具有藥物益處。載體的類別包括但不限于粘合劑、緩沖劑、著色劑、稀釋劑、崩解劑、乳化劑、調味劑、流平劑、潤滑劑、防腐劑、穩定劑、表面活性劑、制片劑、延遲釋放劑和濕潤劑等。稀釋劑可以是惰性稀釋劑,例如碳酸鈣、磷酸鈣或乳糖。崩解劑可以是玉米淀粉或海藻酸。粘合劑可以是淀粉或明膠。潤滑劑可以是硬脂酸鎂、滑石、硬脂酸和十二烷基硫酸鈉。延遲釋放劑可以是例如羧甲基纖維素、鄰苯二甲酸乙酸纖維素或聚乙酸乙烯酯。調味劑可以是甜味劑,甜味劑,例如糖精、環己烷氨基磺酸鹽、甘油或糖;和味道增強劑,例如香蘭素或橙提取物。防腐劑可以是對羥基苯甲酸酯。乳化劑可以是木質素、亞硫酸廢液、甲基纖維素、淀粉和聚乙烯基吡咯烷酮等。
例示性藥學上可接受的載體包括糖、淀粉、纖維素、粉狀黃蓍膠、麥芽、明膠、滑石粉和植物油。
可將本發明的藥物組合物制成各種適合給藥的劑型,包括但不限于片劑、膠囊、溶液、糖漿、乳液等。
因此,可以本領域周知的各種給藥方式給予本發明的藥物組合物,包括但不限于腸外、皮下、靜脈、肌肉、腹腔內、透皮、口腔、鞘內、顱內、鼻腔或外用途徑給藥。作為替代或并行地,可以通過口服給藥。藥的劑量將根據病人的年齡、健康與體重、并行治療的種類、治療的頻率、以及所需治療效益來決定。
本申請的劑型可用已知的方式制備。例如,由傳統的混合、制粒、制錠、溶解、或冷凍干燥過程制造。制造口服制劑時,可結合固體輔料和活性化合物,選擇性研磨混合物。如果需要或必要時加入適量助劑后,加工顆粒混合物,獲得片劑或錠劑芯。
本申請的試劑可含有前文所述的藥學上可接受的載體。當然,也可含有本領域體外實驗常規的各種載體,例如水或能溶解本申請式i化合物的各種無機或有機溶劑。因此,應理解的是,本申請前文所述的各種用途和方法包括體內和體外用途和方法。
本申請的式i化合物或其藥學上可接受的鹽可與其它已知的抗癌藥物一起施用。這類已知的抗癌藥物包括但不限于:白消安、馬法蘭、苯丁酸氮芥、環磷酰胺、異環磷酰胺、替莫唑胺、苯達莫司汀、順鉑、絲裂霉素c、博萊霉素、卡鉑、喜樹堿、伊立替康、托泊替康、阿霉素、表阿霉素、阿克拉霉素、米托蒽醌、甲基羥基玫瑰樹堿、5-氮雜胞苷、吉西他濱、5-氟尿嘧啶、5-氟-2'-去氧尿苷、氟達拉濱、奈拉濱、長春新堿、長春瑞濱、紫杉醇、伊沙匹隆、卡巴他賽、多西他賽、帕尼單抗、阿瓦斯丁、赫賽汀、美羅華、伊馬替尼、吉非替尼、埃羅替尼、拉帕替尼、甲苯磺酸索拉非尼、舒尼替尼、尼羅替尼、達沙替尼、帕唑帕尼、依維莫司、他莫昔芬、來曲唑、氟維司群和米托胍腙等。尤其優選的是,本申請式i化合物或其藥學上可接受的鹽可與甲苯磺酸索拉非尼一起施用。
當使用已知的抗癌藥物時,這類已知的抗癌藥物可以其常規的用量給予,或者以較低的劑量給予。
因此,在某些實施方案中,本申請還包括本申請式i化合物與已知抗癌藥物(尤其是治療肝癌的藥物)在制備治療或預防癌癥(尤其是肝癌)的藥物中的應用。
在其它實施方案中,本申請提供一種藥物組合物,該藥物組合物含有本申請式i化合物或其藥學上可接受的鹽與一種或多種已知的抗癌藥物。所述藥物組合物中本申請式i化合物或其藥學上可接受的鹽與已知的抗癌藥物分別提供,或以兩者的混合物的形式提供。藥物組合物中,所述已知的抗癌藥物的劑量可為該抗癌藥物常規使用時的劑量,或者可以較低的劑量使用或含于本申請的藥物組合物中。在優選的實施方案中,所述抗癌藥物是治療肝癌的藥物,例如甲苯磺酸索拉非尼。在某些實施方案中,該藥物組合物中,甲苯磺酸索拉非尼的單位劑量要低于其常規的用于治療肝癌時的單位劑量。
本申請的藥物組合物可以給予任何哺乳動物對象,優選的,所述哺乳動物是人。
下列實施例是舉例說明,而不是限制本發明的方法和制劑。其他對于本領域技術人員來說是顯而易見的,和在臨床治療中通常會遇到的對各種條件和參數的適當修改和改進,都在本發明的精神和范圍內。
實施例1:化合物747的制備
將長苞冷杉干燥的地上部分22kg粉碎成粗粉,用200l的80%乙醇回流提取3次,每次3小時,合并提取液,減壓濃縮成25l稠浸膏。浸膏加10l水稀釋后分別以氯仿、乙酸乙酯和正丁醇依次萃取,收集各萃取部分,分別濃縮成浸膏。其中乙酸乙酯萃取部分的浸膏300g,以甲醇溶解過濾后進行硅膠柱色譜,以氯仿-甲醇2:1洗脫,用薄層色譜檢查洗脫流分,具有相同單一斑點的流分合并,濃縮,得流分1-10,其中第1流分經進一步硅膠色譜純化,得到黃色無定形粉末,結構鑒定為stenopalustrosided,即山奈酚3-(2,4-二-e-p-香豆酰基鼠李糖苷),共58mg。該化合物結構如下所示,本文命名為化合物747:
化合物747的分子量為724,黃色粉末,極易溶于甲醇、乙醇,易溶于乙酸乙酯、dmso,不溶于水。化合物747的hplc-uv檢測和ms(+/-)分別如圖23的a和b所示。
核磁數據:
無定形粉末;1hnmr(cd3od,600mhz)δ7.76(2h,d,j=9.0hz,h-2′,6′),7.66(1h,d,j=15.6hz,h-7′″),7.57(1h,d,j=15.6hz,h-7″″),7.48(2h,d,j=8.4hz,h-2′″,6′″),7.43(2h,d,j=9.0hz,h-2″″,6″″),7.02(2h,d,j=8.4hz,h-3′,5′),6.81(2h,d,j=8.4hz,h-3′″,5′″),6.78(2h,d,j=8.0hz,h-3′″,5′″),6.38(1h,d,j=15.6hz,h-8′″),6.32(1h,d,j=1.8hz,h-8),6.29(1h,d,j=15.6hz,h-8″″),6.17(1h,d,j=1.8hz,h-6),5.74(1h,d,j=1.8hz,h-1″),5.61(1h,m,h-2″),5.01(1h,t,j=9.6hz,h-4″),4.22(1h,dd,j=9.6,3.6hz,h-3″),3.33(1h,m,h-5″),0.88(3h,d,j=6.0hz,h3-6″);
13cnmr(cdcl3,150mhz),見表1;
esims(positive)m/z747[m+na]+;esims(negative)m/z723[m-h]-,759[m+cl]-。
表1:化合物747的13cnmr光譜數據
實施例2:化合物747的活性測試
1.實驗步驟
1.1細胞遷移
人單核細胞系thp-1(ccr2高表達),96孔transwell(corning3788),hccl2(peprotech),thp-1細胞與藥物共孵育30分鐘后,取1x105thp-1細胞/孔,下層培養基加入hccl250ng/ml誘導其遷移,6小時后
1.2鈣流實驗
thp-1細胞穩轉鈣流指示蛋白gcamp6獲得穩轉株thp-1gcamp6,thp-1細胞與藥物共孵育30分鐘后,取1x106thp-1細胞/孔并加入mg2+:2mm,ca2+:2mm,hccl2100ng/ml(peprotech)記錄數據(488nm,520nm)。
293ft細胞穩轉鈣流指示蛋白gcamp6獲得穩轉株293ft-gcamp6,lipo2000瞬轉pcdnatm3.1/v5-hisa-hccr2,48h后,與藥物共孵育37度,3h,取1x106細胞/孔,加入mg2+:2mm,ca2+:2mm,hccl2100ng/ml(peprotech),記錄數據(488nm,520nm)。
1.3同位素實驗
pcdnatm3.1/v5-hisa-hccr2;hccr5;mccr2;mccr5(h:hμman,m:mouse);lipo2000瞬轉96孔板chok1.1,3500細胞/孔,同位素配體[125i]mcp-1(nex332,pe),[125i]m-ip-1a(nex298,pe)與藥物和細胞共孵育3小時后,加閃爍液,上機記錄數據。
1.4腹膜炎實驗
4%巰基乙酸鹽(sigma),6-8周雄性小鼠腹腔注射1ml,藥物組每天給予相應劑量的腹腔注射,對照組每天打等量的注射液。3天后取腹水細胞,記數,流式分析。
1.5小鼠肝癌腫瘤治療實驗與肝癌微環境細胞
小鼠與人肝癌細胞系hepa1-6、lpc,7404,7702,hepg2等實驗細胞系均來自上海生科院細胞庫或atcc,c57/bl6,balb/c裸鼠(上海斯萊克公司)飼養與spf級動物房內。皮下腫瘤模型接種4周c57/bl6雄性小鼠腹股溝150μl1.5x106細胞/只,腫瘤生長至100mm3左右大小進行分組給藥治療,并追蹤腫瘤大小與小鼠體重。巨噬細胞清除劑氯膦酸二鈉脂質體(formμmax)清除小鼠體內巨噬細胞,每三天腹腔1次,每次150μl/只。cd8中和性抗體(ebioscience)清除小鼠體內cd8t細胞,每三天1次,每次150μl/只。
原位腫瘤模型將hepa1-6穩定標記熒光素酶后,在c57/bl6小鼠肝臟原位注射100μl1x106細胞,xenogen活體成像系統(perkin-elmer)使用前小鼠注射d-luciferin(alameda,ca,usa)用于荷瘤小鼠活體成像,分組給藥治療并追蹤腫瘤進展。所有動物實驗用藥均由0.5%羥甲基纖維素與0.2%tween-20配置。新鮮的小鼠腫瘤組織分離消化成單細胞(miltenyibiotec),紅細胞裂解液去除血紅細胞,重懸于facs溶液并抗鼠cd16/32去除非特異性結合,流式抗體對細胞進行染色(4度避光,45分鐘),bdariaii上機檢測并記錄。實驗所使用的流式抗體以及溶液購自ebioscience,biolegend,r&d公司。
2.實驗結果
在體外實驗中,圖1顯示本發明中747藥物能夠抑制ccl2-ccr2介導的thp-1遷移能力,并呈現一定的劑量依賴。
圖2顯示,與現有的ccr2拮抗劑rdc和rs相比,747藥物在同濃度10μm能夠明顯抑制ccl2-ccr2引起的鈣流。
圖3顯示,747在濃度低至100nm依舊展現很好的抑制鈣流能力。
由于ccr2與ccr5序列有接近70%相似性,因此是現在開發ccr2專一性拮抗劑的一個難點。但圖4和圖5顯示,在同位素的結合抑制實驗中,本發明中的747藥物對于ccl2-人ccr2,ccl2-鼠ccr2的半數抑制濃度分別為167.2nm與177.8nm,而對于人ccr5的半數抑制濃度為16547nm,對鼠ccr5并沒有展現出抑制能力,這說明747藥物能夠直接抑制ccr2這一受體并發揮功能,抑制ccr2介導的細胞遷移以及鈣流,而對于ccr5的抑制能力則很差,展現了很好的選擇性以及敏感性。
體內實驗中,圖6顯示在ccr2介導腹膜炎的巨噬細胞招募的模型中,747也展現了良好的抑制巨噬細胞招募的能力,并呈現一定的劑量依賴。
以上的實驗結果說明在體內外747藥物都對ccr2有良好的拮抗作用,并能抑制ccr2相關功能,具有很好的選擇性以及敏感性的一類新的ccr2拮抗劑。
ccl2-ccr2這一信號通路在肝臟炎癥-肝癌的過程被激活并與肝癌的預后也有一定關系。圖7顯示747對于肝癌細胞系在體外并無明顯的細胞毒殺傷作用,而在圖8和圖9的c57/bl6小鼠的hepa1-6原位肝癌模型中展現出了很好的抑制腫瘤能力,圖11中顯示腫瘤微環境中殺傷性cd8t細胞明顯增多,cd4t細胞沒有明顯變化。因為ccr2主要表達在巨噬細胞里,少量表達于其他細胞,巨噬細胞在腫瘤微環境中通常發揮免疫抑制功能來促進腫瘤,我們認為這一現象表明,747藥物發揮抑制腫瘤的能力不是直接殺傷腫瘤,而是通過抑制ccr2這一靶點來解除微環境中腫瘤巨噬細胞的免疫抑制能力,調動激活cd8t細胞來完成。圖10中相對于sorafenib組小鼠體重沒有降低,表示747藥物該劑量下并沒有明顯毒性。
為了研究747藥物是否通過解除巨噬細胞對cd8t細胞的抑制來發揮抗肝癌的功能,分別使用了巨噬細胞清除劑和cd8中和性抗體來進行研究,在圖12中,747治療組與巨噬細胞清除組展現了相似的腫瘤抑制能力,并且在巨噬細胞清除組747沒有明顯治療效果,并且這一治療效果依賴cd8t細胞,cd8t細胞清除后,治療效果隨即消失。圖13為各治療組對hepa1-6小鼠肝癌皮下腫瘤生長變化測量追蹤,圖14和圖15顯示747治療后hepa1-6小鼠肝癌皮下腫瘤微環境中cd8t細胞顯著增多,巨噬細胞tam顯著減少。
為了證實747對于肝癌的抑制能力并非hepa1-6細胞系依賴,采用了另一株小鼠肝癌細胞系lpc,構建皮下腫瘤治療模型,并與現有肝癌臨床一線用藥sorafenib(甲苯磺酸索拉非尼,商品名:多吉美)聯合使用,采用腫瘤靶向加免疫治療雙管齊下,圖17為747,低劑量sorafenib,與747聯合sorafenib分別治療lpc小鼠皮下肝癌的結果,圖17和圖18分別為各組治療過程腫瘤生長過程的追蹤以及腫瘤最終重量。lpc皮下肝癌治療結果顯示低劑量的sorafenib抑制lpc皮下肝癌能力有限,與對照組無明顯差異,747具有一定的治療效果,兩種藥物聯合使用后,能夠顯著抑制腫瘤生長,并且與對照組與747、sorafenib單獨治療組都有明顯差異。圖19和圖20與顯示747治療lpc小鼠皮下腫瘤后,腫瘤微環境中cd8t細胞顯著增多,cd4t細胞無明顯變化,巨噬細胞tam顯著減少,這一結果與hepa1-6皮下腫瘤結果相似。在圖21中,747治療以后ccr2陽性的細胞出現了顯著的減少,與小鼠腹膜炎模型互相印證,說明了747在體內具有良好的ccr2拮抗活性,此外本發明發現747治療以后微環境中的巨噬細胞數量明顯減少后激活cd8t細胞殺傷腫瘤的同時,巨噬細胞的表型也有一定變化,圖22顯示從m2-免疫抑制型向m1-免疫促進型偏移,協助抑制殺傷腫瘤。
本發明開發了一種天然產物來源的新型的ccr2拮抗劑(實驗代號747),通過各種模型認證其對于ccr2的體內體外的拮抗活性以及敏感性和選擇性,本發明通過ccr2這一免疫靶點調節了腫瘤微環境,減少腫瘤巨噬細胞的浸潤,增加了cd8t細胞達到治療腫瘤的效果,使用本發明可以產生新的骨架結構的ccr2拮抗劑,可用于肝癌的免疫治療。本化合物來源于天然產物本身毒性很低,聯合現有肝癌一線臨床用藥sorafenib,利用小鼠肝癌模型進行聯合治療,利用低劑量sorafenib聯合本化合物輔助免疫治療展現了很好的治療效果,對于現在臨床的耐藥以及高劑量抗腫瘤藥帶來毒副作用提供了新的臨床應用前景,為臨床治療提供新的思路。
- 還沒有人留言評論。精彩留言會獲得點贊!