HER2抗體?藥物綴合物的制作方法
本申請根據35U.S.C.§119(e)要求2014年6月23日提交的美國臨時專利申請U.S.S.N.62/015,661和2014年6月20日提交的U.S.S.N.62/014,912的優先權,其各自在此引入作為參考。
發明領域
本發明屬于抗癌治療的領域,且通過抗體-藥物綴合物(ADC)形式,尤其對癌細胞,提供細胞毒性藥物遞送的功效和特異性。
背景技術:
抗體-藥物綴合物(ADC)為將單克隆抗體(mAbs)的特異性與細胞毒性分子的效能組合的一類治療劑。ADC的使用通過綴合的細胞毒性劑而賦予抗體癌癥殺傷活性,同時靶特異性遞送避免由暴露于游離毒性劑而造成的全身毒性。目前,兩種ADC已經FDA批準而用于治療人類癌癥。(Brentuximab vedotin或SGN-35),一種與細胞毒性劑MMAE綴合的抗CD30抗體,設計用于治療CD30-陽性復發性淋巴瘤。(T-DM1),一種與細胞毒性劑DM1綴合的抗-HER2抗體,設計用于治療HER2-陽性轉移性乳腺癌。
連接基技術深刻影響ADC效力、特異性和安全性。酶不穩定連接基利用細胞內部和外部的蛋白酶的差別化活性以實現對藥物釋放的控制。藥物可以通過肽鍵與抗體綴合,并且僅可以通過存在于細胞內的溶酶體蛋白酶的作用特異性斷裂,并且在某些腫瘤類型中以升高的水平被特異性斷裂(Koblinsk等人,2000)。這將確保連接基在血流中的穩定性,以限制對健康組織的損傷。然而,一些酶不穩定的連接基的增加的相關疏水性可導致ADC的聚集,特別是對于強疏水性藥物。因此,需要能夠提供血清穩定性以及增加的溶解性的連接基,允許疏水性藥物的有效綴合和細胞內遞送。
人表皮生長因子受體2蛋白,HER2(ErbB2)是表皮生長因子受體家族的成員。已知這些受體酪氨酸激酶在發育和腫瘤發生中都起關鍵作用。在25%-30%的原發性乳腺癌中觀察到HER2蛋白的過表達(Press等人,1993),因此成為癌靶向治療的重要候選物。已經在體外和在小鼠異種移植模型中顯示人源化抗HER2抗體曲妥珠單抗抑制過表達HER2的人腫瘤細胞的增殖(Hudziak等人,1989;Baselga等人,1998)。雖然曲妥珠單抗是臨床上有活性的并且在治療患有HER2過表達轉移性乳腺癌的患者中有效(Baselga等,1996),但最初對曲妥珠單抗應答的大多數人在一年內產生抗性(Romond等,2005;Nahta等,2006;Pohlmann等,2009)。因此,需要開發針對過表達HER2的腫瘤細胞的新療法。
技術實現要素:
本發明的化合物包含藥物部分、能靶向所選擇的細胞群的靶向部分、和包含酰基單元的連接基、用于提供在藥物部分和靶向部分之間的距離的任選的間隔基單元、可在合適的條件下斷裂的肽連接基、親水性自降解(self-immolative)連接基、和任選的第二自降解間隔基或環化自消除型連接基。
本發明還提供式(I)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
X為親水性自降解連接基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
本發明還提供式(II)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
在一些實施方案中,提供式(Ia)的化合物:
或其鹽或溶劑合物或立體異構體;其中D、T、X、L1、L2、L3、L4和A如式(I)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在一些實施方案中,提供式(IIa)的化合物:
或其鹽或溶劑合物或立體異構體;其中D、T、L1、L2、L3、L4和A如式(II)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在式I、II、Ia和IIa的化合物的一些實施方案中,T為抗體靶向分子。在一些實施方案中,T為抗HER2抗體。在一些實施方案中,T為單克隆抗HER2抗體。在一些實施方案中所述單克隆抗HER2抗體為帕妥珠單抗。在一些實施方案中,所述單克隆抗HER2抗體為margetuximab。在一些實施方案中,T為人源化抗HER2抗體。在一些實施方案中,所述人源化抗HER2抗體為曲妥珠單抗。
在式I、II、Ia和IIa的化合物的一些實施方案中,該抗體的重鏈和/或輕鏈的一個或多個氨基酸殘基被半胱氨酸殘基代替。在一些實施方案中,該抗體的Fc區的一個或多個氨基酸殘基被半胱氨酸殘基代替。在一些實施方案中,該抗體的一個或多個氨基酸殘基在輕鏈的位置147、188、200、201和/或206,和/或在該重鏈的位置155、157、165、169、197、199、209、211和/或442被半胱氨酸殘基代替,使用EU編號(Kabat中的EU索引)。在一些實施方案中,D通過半胱氨酸殘基連接至T。在一些實施方案中,D為含氨基的藥物部分,其中該藥物通過含氨基的藥物部分的氨基連接至L1或X。在一些實施方案中,D為多卡霉素、多拉司他汀、tubulysin、多柔比星(DOX)、紫杉醇或絲裂霉素C(MMC),或它們的氨基衍生物。
在上述實施方案任一個中,A-L4-L3-L2-X-L1-D為:
本發明一些方面涉及藥物組合物,其包含本文所述的化合物或其鹽或溶劑合物或立體異構體;和藥學上可接受的載體。
本發明一些方面涉及殺死細胞的方法。該方法包括向細胞給藥足以殺死細胞的量的本文所述的化合物或其鹽或溶劑合物或立體異構體或藥物組合物。在一些實施方案中,所述細胞為癌細胞。在一些實施方案中,所述癌細胞為乳腺癌細胞、胃癌細胞或卵巢癌細胞。
本發明一些方面涉及在需要的個體中治療癌癥的方法。該方法包括向個體給藥有效量的本文所述的化合物或其鹽或溶劑合物或立體異構體或藥物組合物。在一些實施方案中,所述癌癥為乳腺癌、胃癌或卵巢癌。
本發明一些方面涉及式I、Ia、II或Iia的化合物或其鹽或溶劑合物或立體異構體或藥物組合物,其用于治療癌癥。在一些實施方案中,所述癌癥為乳腺癌、胃癌或卵巢癌。
在一些實施方案中,L1為鍵。在一些實施方案中,L1為自降解連接基。在一些實施方案中,L1為氨基芐基氧基羰基連接基。在一些實施方案中,
L1選自
其中n為1或2。
在一些實施方案中,L1選自
在一些實施方案中,L2為鍵。在一些實施方案中,L2為自降解連接基。在一些實施方案中,L2為氨基芐基氧基羰基連接基。在一些實施方案中,L2選自
其中n為1或2。
在一些實施方案中,L3為具有1至10個氨基酸殘基的肽連接基。在一些實施方案中,L3為具有2至4個氨基酸殘基的肽連接基。在一些實施方案中,L3為具有1、2、3、4、5、6、7、8、9或10個氨基酸殘基的肽連接基。在一些實施方案中,L3為包含至少一個賴氨酸或至少一個精氨酸殘基的肽連接基。在一些實施方案中,L3為包含選自以下的氨基酸殘基的肽連接基:賴氨酸、D-賴氨酸、瓜氨酸、精氨酸、脯氨酸、組氨酸、鳥氨酸和谷氨酰胺。在一些實施方案中,L3為包含選自以下的氨基酸殘基的肽連接基:纈氨酸、異亮氨酸、苯丙氨酸、蛋氨酸、天冬酰胺、脯氨酸、丙氨酸、亮氨酸、色氨酸和酪氨酸。在一些實施方案中,L3為選自以下的二肽單元:纈氨酸-瓜氨酸、脯氨酸-賴氨酸、蛋氨酸-D-賴氨酸、天冬酰胺-D-賴氨酸、異亮氨酸-脯氨酸、苯丙氨酸-賴氨酸和纈氨酸-賴氨酸。在一些實施方案中,L3為纈氨酸-瓜氨酸。
在一些實施方案中,L4為鍵。在一些實施方案中,L4為間隔基。在一些實施方案中,所述間隔基為聚亞烷基二醇、亞烷基、亞烯基、亞炔基、或聚胺。在一些實施方案中,L4為L4a-C(O),L4a-C(O)-NH,L4a-S(O)2,或L4a-S(O)2-NH,其中各L4a獨立地為聚亞烷基二醇、亞烷基、亞烯基、亞炔基、或聚胺。在一些實施方案中,L4為L4a-C(O),其中L4a為聚亞烷基二醇、亞烷基、亞烯基、亞炔基、或聚胺。在一些實施方案中,L4為L4a-C(O),其中L4a為聚亞烷基二醇。在一些實施方案中,L4為L4a-C(O),其中L4a為聚乙二醇。在一些實施方案中,所述間隔基為式-CH2-(CH2-O-CH2)m-CH2-C(O)-,其中m為0至30的整數。在一些實施方案中,所述間隔基為式-CH2-(CH2-O-CH2)m-CH2-C(O)-,其中m為整數1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30。在一些實施方案中,L4為L4a-C(O),其中L4a為亞烷基。
在一些實施方案中,A選自
其中各Q2為NH或O,且各q為整數1至10,且各q1獨立地為1至10的整數。在一些實施方案中,q為2、3、4或5。在某些實施方案中,q1為2、3、4或5。在其它實施方案中,各q為整數1、2、3、4、5、6、7、8、9或10。在一些實施方案中,A為
其中各Q2獨立地為NH或O且各q獨立地為1至10的整數。在其它實施方案中,各q獨立地為整數1、2、3、4、5、6、7、8、9或10。在一些實施方案中,q為2、3、4或5。在一些實施方案中,A選自基團
其中各Q2獨立地為NH或O。
在一些實施方案中,D為含氨基的藥物部分,其中該藥物通過含氨基的藥物部分的氨基連接至L1或X。在一些實施方案中,D為選自以下的多卡霉素的氨基衍生物:
在一些實施方案中,D為多拉司他汀的氨基衍生物(例如單甲基多拉司他汀10):
在一些實施方案中,A-L4-L3-L2為
在一些實施方案中,A-L4-L3-L2-X-L1-D為:
在一些實施方案中,A-L4-L3-L2-X-L1-D為:
在一些實施方案中,A-L4-L3-L2-X-L1-D為:
本發明一些方面涉及制備式(II)的化合物或其鹽或溶劑合物或立體異構體的方法:
其中:
D為藥物部分;
T為抗體;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。該方法包括將抗體與化合物Z或其鹽或溶劑合物或立體異構體反應:
本發明一些方面涉及制備式(IIa)的化合物或其鹽或溶劑合物或立體異構體的方法:
其中:
p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
D為藥物部分;
T為抗體;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。該方法包括將抗體與化合物Z或其鹽或溶劑合物或立體異構體反應:
在一些實施方案中,該抗體為抗HER2抗體。在一些實施方案中,該抗體為單克隆抗HER2抗體。在一些實施方案中,該抗體為人源化抗HER2抗體,任選為單克隆人源化抗HER2抗體。在一些實施方案中,該抗體重鏈和/或輕鏈的一個或多個氨基酸殘基被半胱氨酸殘基代替。在一些實施方案中,該抗體的Fc區的一個或多個氨基酸殘基被半胱氨酸殘基代替。在一些實施方案中,在輕鏈的位置147、188、200、201和/或206,和/或在重鏈的位置155、157、165、169、197、199、209、211和/或442,該抗體的一個或多個氨基酸殘基被半胱氨酸殘基代替,使用EU編號(Kabat中的EU索引)。在一些實施方案中,該抗體包括一個或多個巰基。在一些實施方案中,該化合物使用本文所述的方法之一制備,其中所述抗體包括一個或多個巰基。
還提供了藥物組合物,其包含本文所述的化合物或其鹽或溶劑合物或立體異構體,和藥學上可接受的載體。
本發明一些方面涉及式(IX)的化合物
或其鹽或溶劑合物或立體異構體;其中R為NO2或NH2.
本發明一些方面涉及制備化合物X或其鹽或溶劑合物或立體異構體的方法:
其中:
L2為鍵或自降解連接基;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。該方法包括將化合物W:A-L4-L3-L2;與化合物I反應:
本發明一些方面涉及制備化合物Z或其鹽或溶劑合物或立體異構體的方法:
其中:
D為藥物部分;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
該方法包括:將化合物X:和氯甲酸對硝基苯酯反應以形成化合物Y:
和將化合物Y與包含L1-D的化合物反應。
本發明一些方面涉及制備化合物X1或其鹽或溶劑合物或立體異構體的方法:
其中:
L2為鍵或自降解連接基;
L3為肽連接基;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。該方法包括:將化合物W1:L3-L2;和化合物I反應:
本發明一些方面涉及制備化合物Y1或其鹽或溶劑合物或立體異構體的方法:
其中:
D為藥物部分;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。該方法包括:在氯甲酸對硝基苯酯的存在下將化合物X1:
和包含L1-D的化合物反應。在一些實施方案中,在制備化合物Y1的方法中,將選自二(4/對硝基苯基)碳酸酯、光氣、三光氣/雙(三氯甲基碳酸酯)、氯甲酸三氯甲酯、N,N’-二琥珀酰亞胺基碳酸酯和1,1’-羰基二咪唑的化合物,代替氯甲酸對硝基苯酯。
本發明一些方面涉及制備化合物Z或其鹽或溶劑合物或立體異構體的方法:
其中:
D為藥物部分;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;
A為酰基單元;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
該方法包括:將化合物Y1:
和包含A-L4的化合物反應。
還提供了下式的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
L2為鍵或自降解連接基;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
還提供了下式的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
還提供了下式的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
L2為鍵或自降解連接基;
L3為肽連接基;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
還提供了下式的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;且
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
本發明還提供式(III)的化合物:
或其鹽或溶劑合物或立體異構體;NH
其中T為靶向部分。
在一些實施方案中,提供了式(IIIa)的化合物:
或其鹽或溶劑合物或立體異構體;其中T為靶向部分且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(IV)的化合物:
或其鹽或溶劑合物或立體異構體;
其中T為靶向部分。
在一些實施方案中,提供了式(IVa)的化合物:
或其鹽或溶劑合物或立體異構體;其中T為靶向部分且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(V)的化合物:
或其鹽或溶劑合物或立體異構體;
其中T為靶向部分。
在一些實施方案中,提供了式(Va)的化合物:
或其鹽或溶劑合物或立體異構體;其中T為靶向部分且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(VI)的化合物:
或其鹽或溶劑合物。
本發明提供式(VII)的化合物:
或其鹽或溶劑合物。
本發明提供式(VIII)的化合物:
本發明提供式(IX)的化合物:
或其鹽或溶劑合物或立體異構體;其中R為NO2或NH2。
在某些實施方案中,式(I)、(II)、(III)、(IV)、(V)、(VI)、(VII)、(VIII)或(IX)的化合物為選自本文發明詳述中描述或示例的那些物質的化合物。
在式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物的某些實施方案中,T為抗體靶向分子。在一些其它實施方案中,T為抗HER2抗體。在其它實施方案中,該抗HER2抗體為人源化或單克隆或人源化單克隆抗體。在一些實施方案中,T為人源化單克隆抗HER2抗體曲妥珠單抗。在一些實施方案中該單克隆抗HER2抗體為帕妥珠單抗。在一些實施方案中,該單克隆抗HER2抗體為margetuximab。
在其它實施方案中,該抗體的重鏈和/或輕鏈的一個或多個氨基酸殘基被半胱氨酸殘基代替(例如,工程化以在不存在于母體抗體中的位置包含半胱氨酸殘基)。在一些實施方案中,該抗體的Fc區的一個或多個氨基酸殘基被半胱氨酸殘基代替。在一些實施方案中,該抗體的一個或多個氨基酸殘基在該輕鏈的位置147、188、200、201和/或206,和/或在該重鏈的位置155、157、165、169、197、199、209、211和/或442,使用EU編號(Kabat中的EU索引)。在一些實施方案中,包含工程化半胱氨酸殘基的抗體為抗HER2抗體。在式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物的一些實施方案中,D通過半胱氨酸(例如工程化的)殘基連接至T。
在某些實施方案中,D為含氨基的藥物部分。在一些實施方案中,D通過氨基連接至L1或X。在其它實施方案中,D為多卡霉素、多拉司他汀、tubulysin、多柔比星(DOX)、紫杉醇或絲裂霉素C(MMC),或它們的氨基衍生物。
在另一方面,本發明提供藥物組合物,其包含至少一種式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其藥物可接受的鹽。根據該實施方案的藥物組合物還可包含藥物可接受的賦形劑。本發明還提供式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其藥物可接受的鹽,其用作藥物。
在另一方面,本發明提供殺死細胞的方法,包括向細胞給藥足以殺死細胞的量的式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物。在一些實施方案中,所述細胞為癌細胞。在其它實施方案中,所述癌細胞為乳腺癌細胞、胃癌細胞或卵巢癌細胞。
在另一方面,本發明提供在需要的個體中治療癌癥的方法,包括向該個體給藥有效量的式(I)-(V)或(Ia)-(Va)的化合物或其鹽、溶劑合物或立體異構體。可用本文所述的方法治療的癌癥的實例包括,但不限于,乳腺、膀胱、胰腺、非小細胞非癌(NSCLC)、卵巢、子宮內膜、結腸、腎、頭與頸、胃、食管、前列腺、和睪丸生殖細胞的癌癥,子宮癌,維爾姆斯腫瘤。在一些實施方案中,在式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物中,T為抗HER2抗體且D為含氨基的藥物部分。在其它實施方案中,T為抗體曲妥珠單抗且D為單甲基多拉司他汀10。在一些實施方案中,T為抗體帕妥珠單抗且D為單甲基多拉司他汀10。在一些實施方案中,T為抗體margetuximab且D為單甲基多拉司他汀10。
在另一方面,本發明提供式(I)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
X為親水性自降解連接基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元,
其用于治療癌癥。
在另一方面,本發明提供式(II)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元,
其用于治療癌癥。
在另一方面,本發明提供式(II)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元,
其用于治療癌癥。
在另一方面,本發明提供式(Ia)的化合物:
或其鹽或溶劑合物或立體異構體;其中D、T、X、L1、L2、L3、L4和A如式(I)所定義,且p為1至20,其用于治療癌癥。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在一些實施方案中,提供式(IIa)的化合物:
或其鹽或溶劑合物或立體異構體;其中D、T、L1、L2、L3、L4和A如式(II)所定義,且p為1至20,其用于治療癌癥。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在一些方面,本發明還提供式(III)的化合物:
或其鹽或溶劑合物或立體異構體;
其中T為靶向部分,其用于治療癌癥。
在一些實施方案中,提供了式(IIIa)的化合物:
或其鹽或溶劑合物或立體異構體;其中T為靶向部分且p為1至20,其用于治療癌癥。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(IV)的化合物:
或其鹽或溶劑合物或立體異構體;
其中T為靶向部分,其用于治療癌癥。
在一些實施方案中,提供了式(IVa)的化合物:
或其鹽或溶劑合物或立體異構體;其中T為靶向部分且p為1至20,其用于治療癌癥。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(V)的化合物:
或其鹽或溶劑合物或立體異構體;
其中T為靶向部分,其用于治療癌癥。
在一些實施方案中,提供了式(Va)的化合物:
或其鹽或溶劑合物或立體異構體;其中T為靶向部分且p為1至20,其用于治療癌癥。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在另一方面,本發明提供藥物組合物,其包含至少一種式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其藥物可接受的鹽,其用于治療癌癥。根據該實施方案的藥物組合物還可包含藥物可接受的賦形劑。本發明還提供式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其藥物可接受的鹽,其用作藥物。
在另一方面,本發明提供式(I)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中:
D為藥物部分;
T為靶向部分;
X為親水性自降解連接基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
在另一方面,本發明提供式(II)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
在另一方面,本發明提供式(II)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
在另一方面,本發明提供式(Ia)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中D、T、X、L1、L2、L3、L4和A如式(I)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在一些實施方案中,提供式(IIa)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中D、T、L1、L2、L3、L4和A如式(II)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在一些方面,本發明還提供式(III)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中T為靶向部分。
在一些實施方案中,提供式(IIIa)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中T為靶向部分且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(IV)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中T為靶向部分。
在一些實施方案中,提供式(IVa)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中T為靶向部分且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明提供式(V)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中T為靶向部分。
在一些實施方案中,提供式(Va)的化合物或其鹽或溶劑合物或立體異構體在制備用于治療癌癥的藥物中的用途:
其中T為靶向部分且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
在一些方面,本發明提供藥物組合物(其包含至少一種式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其藥物可接受的鹽)在制備用于治療癌癥的藥物中的用途。根據該實施方案的藥物組合物還可包含藥物可接受的賦形劑。本發明還提供式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其藥物可接受的鹽,其用作藥物。
在以上討論的任一實施方案中,所述抗HER2抗體包含重鏈可變區和輕鏈可變區,其中
(1)該重鏈可變區包含SEQ ID NO:16-18的氨基酸序列的三個重鏈CDR和/或該輕鏈可變區包含SEQ ID NO:19-21的氨基酸序列的三個輕鏈CDR;
(2)該重鏈可變區包含SEQ ID NO:22-24的氨基酸序列的三個重鏈CDR和/或該輕鏈可變區包含SEQ ID NO:25-27的氨基酸序列的三個輕鏈CDR;或
(3)該重鏈可變區包含SEQ ID NO:28-30的氨基酸序列的三個重鏈CDR和/或該輕鏈可變區包含SEQ ID NO:31-33的氨基酸序列的三個輕鏈CDR。
在以上討論的任一實施方案中,該抗HER2抗體包含重鏈可變區和輕鏈可變區,其中
(1)該重鏈可變區包含SEQ ID NO:8的氨基酸序列和/或該輕鏈可變區包含SEQ ID NO:7的氨基酸序列;
(2)該重鏈可變區包含SEQ ID NO:13的氨基酸序列和/或該輕鏈可變區包含SEQ ID NO:12的氨基酸序列;
(3)該重鏈可變區包含SEQ ID NO:15的氨基酸序列和/或該輕鏈可變區包含SEQ ID NO:14的氨基酸序列。
本發明的其它實施方案、特征和優點根據以下發明詳述和通過本發明的實踐將是明顯的。
附圖簡述
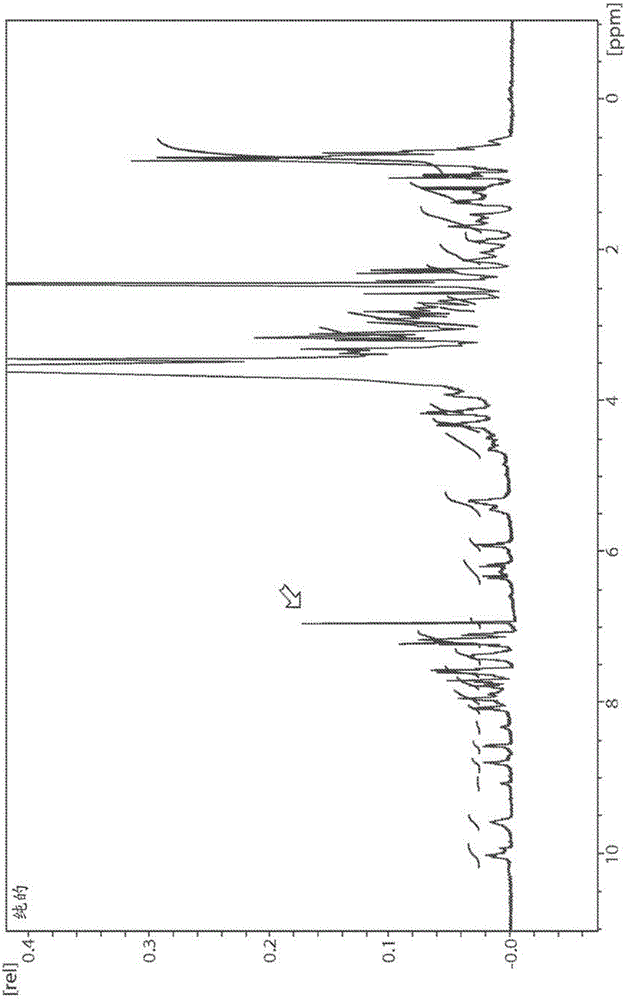
圖1顯示Tap-18H的NMR譜。
圖2顯示Tap-18Hr1的NMR譜。
圖3顯示Tap-18Hr2的NMR譜。
圖4顯示抗HER2-IgG1/TAP18Hr1對抗卵巢癌SKOV-3的體內抗腫瘤活性。
圖5顯示抗HER2-IgG1/TAP18Hr1對抗乳腺癌MDA-MB-453的體內抗腫瘤活性。箭頭指示ADC治療的時間(第1天)。
圖6顯示抗HER2-IgG1/TAP18Hr1對抗胃癌NCI-N87的體內抗腫瘤活性。箭頭指示ADC治療的時間(第1天和第22天)。
圖7顯示位點特異的綴合的抗HER2-Cys變體對抗胃癌NCI-N87的體內抗腫瘤活性。箭頭指示ADC治療的時間(第1天)。
圖8顯示Tap18Hr1常規綴合的抗HER2和位點特異的綴合的抗HER2-Cys變體對抗乳腺癌JIMT-1的體內抗腫瘤活性。箭頭指示ADC治療的時間(第1天)。
發明詳述
本公開提供了具有親水性自降解連接基的化合物,其在合適的條件下是可斷裂的并且引入親水基團以提供化合物的更好的溶解性。對于通常為疏水的細胞毒性藥物,該親水性自降解連接基可提供藥物綴合物增加的溶解性。在藥物綴合物中使用親水性自降解連接基的其他優點包括藥物綴合物增加的穩定性和藥物綴合物降低的聚集。
本公開提供了可以具有優異的血清穩定性的藥物綴合物。例如,與其中藥物的羥基通過易于在水性緩沖液或人血清中快速水解的不穩定碳酸酯鍵連接到間隔基的藥物綴合物相反,本申請的藥物綴合物利用芐氧基羰基鍵。這些綴合物在相同條件下相對更穩定,并且在用蛋白酶例如組織蛋白酶B處理時選擇性地經歷斷裂以釋放藥物。對于藥物綴合物,血清穩定性是期望的性質,其中期望將惰性藥物施用于患者的血清,通過配體使惰性藥物集中于靶標上,然后使藥物綴合物僅在靶標附近轉化為活性形式。
本公開提供了可以具有減少的聚集的藥物綴合物。一些酶不穩定連接基的增加的相關疏水性可能導致藥物綴合物的聚集,特別是對于強疏水性藥物。通過將親水基團引入連接基中,藥物綴合物的聚集減少。與具有化學不穩定連接基和不可斷裂連接基的ADC相比,本文描述的連接基可以通過特異性酶不穩定設計獲得更好的血清穩定性,以及通過對異質癌細胞的旁觀者效應(bystander effect)實現更好的功效。本發明的化合物包含藥物部分、能靶向所選擇的細胞群的靶向部分、和包含酰基單元的連接基、用于提供在藥物部分和靶向部分之間的距離的任選的間隔基單元、可在合適的條件下斷裂的肽連接基、親水性自降解連接基、和任選的第二自降解間隔基或環化自消除型連接基。下面討論每個特征。
“烷基”是指具有1、2、3、4、5、6、7、8、9或10碳原子且優選1、2、3、4、5或6個碳原子的一價飽和脂族烴基。該術語包括,作為實例,直鏈和支鏈烴基如甲基(CH3-)、乙基(CH3CH2-)、正丙基(CH3CH2CH2-)、異丙基((CH3)2CH-)、正丁基(CH3CH2CH2CH2-)、異丁基((CH3)2CHCH2-)、仲丁基((CH3)(CH3CH2)CH-)、叔丁基((CH3)3C-)、正戊基(CH3CH2CH2CH2CH2-)、新戊基((CH3)3CCH2-)和正己基(CH3(CH2)5-)。
“亞烷基”是指優選具有1、2、3、4、5、6、7、8、9或10且更優選1、2或3個碳原子的二價脂族亞烴基,其為直鏈或支鏈的。該術語包括,作為實例,亞甲基(-CH2-)、亞乙基(-CH2CH2-)、正亞丙基(-CH2CH2CH2-)、異亞丙基(-CH2CH(CH3)-)、(-C(CH3)2CH2CH2-)、(-C(CH3)2CH2C(O)-)、(-C(CH3)2CH2C(O)NH-)、(-CH(CH3)CH2-),等。
“烯基”是指具有2、3、4、5、6、7、8、9或10個碳原子且優選2、3或4個碳原子和具有至少1個且優選1至2個雙鍵位點的直鏈或支鏈烴基。該術語包括,作為實例,二-乙烯基、烯丙基和丁-3-烯-1-基。該術語中包括順式和反式異構體或這些異構體的混合物。
“亞烯基”是指具有2、3、4、5、6、7、8、9或10個碳原子且優選2、3或4個碳原子和具有至少1個且優選1至2個雙鍵不飽和位點的直鏈或支鏈亞烴基。該術語包括,作為實例,二-乙烯基,烯丙基和丁-3-烯-1-基。該術語中包括順式和反式異構體或這些異構體的混合物。
“炔基”是指具有2、3、4、5或6個碳原子且優選2至3個碳原子和具有至少1個且優選1至2個三鍵不飽和位點的直鏈或支鏈烴基。這種炔基的實例包括乙炔基(-C≡CH)和炔丙基(-CH2C≡CH)。
“亞炔基”是指具有2、3、4、5或6個碳原子且優選2至3個碳原子和具有至少1個且優選1至2個三鍵不飽和位點的直鏈或支鏈亞烴基。該炔基的實例包括乙炔基(-C≡CH)和炔丙基(-CH2C≡CH)。
“氨基”是指基團–NH2。
“取代的氨基”是指基團-NRR,其中各R獨立地選自氫、烷基、取代的烷基、環烷基、取代的環烷基、烯基、取代的烯基、環烯基、取代的環烯基、炔基、取代的炔基、芳基、雜芳基和雜環基,條件是至少一個R不為氫。
“芳基”是指6、7、8、9、10、11、12、13、14、15、16、17或18個碳原子的一價芳族碳環基團,其具有單環(如以苯基存在)或有多個稠合的環的環體系(該芳環體系的實例包括萘基、蒽基和茚滿基),該稠合的環可為或可不為芳族的,條件是連接點是經由芳環的原子。該術語包括,作為實例,苯基和萘基。除非另外被芳基取代基的定義所限制,該芳基可任選取代有1、2、3、4或5個取代基,或1、2或3個取代基,所述取代基選自酰基氧基、羥基、巰基、酰基、烷基、烷氧基、烯基、炔基、環烷基、環烯基、取代的烷基、取代的烷氧基、取代的烯基、取代的炔基、取代的環烷基、取代的環烯基、氨基、取代的氨基、氨基酰基、酰基氨基、烷芳基、芳基、芳基氧基、疊氮基、羧基、羧基酯、氰基、鹵素、硝基、雜芳基、雜芳基氧基、雜環基、雜環氧基、氨基酰基氧基、氧基酰基氨基、硫代烷氧基(thioalkoxy)、取代的硫代烷氧基、硫代芳基氧基、硫代雜芳基氧基、磺酰基氨基、-SO-烷基、-SO-取代的烷基、-SO-芳基、-SO-雜芳基、-SO2-烷基、-SO2-取代的烷基、-SO2-芳基、-SO2-雜芳基和三鹵代甲基。
“環烷基”是指3,4,5,6,7,8,9,或10個碳原子的環烷基,其具有單一環或多個環,包括稠合、橋接和螺環體系。合適的環烷基的實例包括,例如,金剛烷基、環丙基、環丁基、環戊基、環辛基等。該環烷基包括,作為實例,單環結構如環丙基、環丁基、環戊基、環辛基、等,或多環結構如金剛烷基,等。
“雜芳基”是指在環中有1、2、3、4、5、6、7、8、9、10、11、12、13、14、或15個碳原子,如1、2、3、4、5、6、7、8、9或10個碳原子和1、2、3、4、5、6、7、8、9或10個選自氧、氮和硫的雜原子的芳族基。該雜芳基可具有單環(如,吡啶基、咪唑基或呋喃基)或在環體系有多個稠合的環(例如像在以下基團中,如,吲嗪基、喹啉基、苯并呋喃、苯并咪唑基或苯并噻吩基),其中環體系中至少一個環為芳族的且環體系中至少一個環為芳族的,條件是連接點是經由芳環的原子。在某些實施方案中,雜芳基的氮和/或硫環原子任選被氧化以提供N-氧化物(N→O)、亞硫酰基或磺酰基部分。該術語包括,作為實例,吡啶基、吡咯基、吲哚基、噻吩基和呋喃基。除非另外被雜芳基取代基的定義所約束,該雜芳基可任選被1、2、3、4或5個取代基,或1、2或3個取代基取代,所述取代基選自酰基氧基、羥基、巰基、酰基、烷基、烷氧基、烯基、炔基、環烷基、環烯基、取代的烷基、取代的烷氧基、取代的烯基、取代的炔基、取代的環烷基、取代的環烯基、氨基、取代的氨基、氨基酰基、酰基氨基、烷芳基、芳基、芳基氧基、疊氮基、羧基、羧基酯、氰基、鹵素、硝基、雜芳基、雜芳基氧基、雜環基、雜環氧基、氨基酰基氧基、氧基酰基氨基、硫代烷氧基、取代的硫代烷氧基、硫代芳基氧基、硫代雜芳基氧基、磺酰基氨基、-SO-烷基、-SO-取代的烷基、-SO-芳基、-SO-雜芳基、-SO2-烷基、-SO2-取代的烷基、-SO2-芳基和-SO2-雜芳基、以及三鹵代甲基。
雜芳基的實例包括,但不限于,吡咯、咪唑、吡唑、吡啶、吡嗪、嘧啶、噠嗪、吲嗪、異吲哚、吲哚、嘌呤、異喹啉、喹啉、酞嗪、萘基吡啶、喹喔啉、喹唑啉、噌啉、蝶啶、咔唑、咔啉、菲啶、吖啶、菲咯啉、異噻唑、吩嗪、異噁唑、吩噁嗪、吩噻嗪、哌啶、哌嗪、鄰苯二甲酰亞胺、4,5,6,7-四氫苯并[b]噻吩、噻唑、噻吩、苯并[b]噻吩,等。
“雜環”、“雜環的”、“雜環烷基”或“雜環基”是指飽和或部分不飽和基團,其具有單環或多個縮合的環,包括稠合、橋接或螺環體系,且具有3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、或20個環原子,包括1、2、3、4、5、6、7、8、9或10個雜原子。這些環原子選自碳、氮、硫或氧,其中,在稠合的環體系中,所述環的一個或多個可為環烷基、芳基或雜芳基,條件是連接點是經由非芳環。在某些實施方案中,雜環基的氮和/或硫原子任選被氧化以提供N-氧化物、-S(O)-或–SO2-部分。
雜環的實例包括,但不限于,氮雜環丁烷、二氫吲哚、吲唑、喹嗪、咪唑烷、咪唑啉、哌啶、哌嗪、二氫吲哚、1,2,3,4-四氫異喹啉、噻唑烷、嗎啉基、硫嗎啉基(也稱為硫代嗎啉基(thiamorpholinyl))、1,1-二氧代硫嗎啉基、哌啶基、吡咯烷、四氫呋喃基,等。
當雜芳基或雜環基是“取代的”時,除非另外被雜芳基或雜環取代基的定義所約束,這些雜芳基或雜環基可取代有1、2、3、4或5個,或1、2或3個取代基,所述取代基選自烷基、取代的烷基、烷氧基、取代的烷氧基、環烷基、取代的環烷基、環烯基、取代的環烯基、酰基、酰基氨基、酰基氧基、氨基、取代的氨基、氨基酰基、氨基酰基氧基、疊氮基、氰基、鹵素、羥基、氧代、硫酮基(thioketo)、羧基、羧基酯、硫代芳基氧基、硫代雜芳基氧基、硫基雜環氧基、巰基、硫代烷氧基、取代的硫代烷氧基、芳基、芳基氧基、雜芳基、雜芳基氧基、雜環基、雜環氧基、羥基氨基、烷氧基氨基、硝基、磺酰基氨基、-SO-烷基、-SO-取代的烷基、-SO-芳基、-SO-雜芳基、-SO-雜環基、-SO2-烷基、-SO2-取代的烷基、-SO2-芳基、-SO2-雜芳基、和-SO2-雜環基。
“聚亞烷基二醇”是指直鏈或支鏈的聚亞烷基二醇聚合物如聚乙二醇、聚丙二醇和聚丁二醇。聚亞烷基二醇亞單元為單一聚亞烷基二醇單元。例如,聚乙二醇亞單元的實例為乙二醇-O-CH2-CH2-O-,或丙二醇-O-CH2-CH2-CH2-O-,在鏈端點以氫封端。其它聚(亞烷基二醇)的實例包括,但不限于,PEG、PEG衍生物如甲氧基聚(乙二醇)(mPEG)、聚(環氧乙烷)、PPG、聚(四亞甲基二醇)、聚(環氧乙烷-共聚-環氧丙烷),或其共聚物和組合。
“聚胺”是指在單體單元中具有胺官能團的聚合物,其或者摻入主鏈,如聚亞烷基亞胺中那樣,或摻入側基,如聚乙烯基胺那樣。
除了本文的公開內容,當用于修飾指定基或基團時,術語“取代的”還可以表示指定基或基團的一個或多個氫原子各自彼此獨立地被如下定義的相同的或不同取代基代替。
除了本文關于單獨術語公開的基團,在指定基或基團中取代飽和碳原子上一個或多個氫(單個碳上的任何兩個氫可被=O、=NR70、=N-OR70、=N2或=S代替)的取代基,除非另有所述,為-R60、鹵素、=O、-OR70、-SR70、-NR80R80、三鹵代甲基、-CN、-OCN、-SCN、-NO、-NO2、=N2、-N3、-S(O)R70、-SO2R70、-SO2O–M+、-SO2OR70、-OSO2R70、-OSO2O–M+、-OSO2OR70、-P(O)(O–)2(M+)2、-P(O)(OR70)O–M+、-P(O)(OR70)2、-C(O)R70、-C(S)R70、-C(NR70)R70、-C(O)O–M+、-C(O)OR70、-C(S)OR70、-C(O)NR80R80、-C(NR70)NR80R80、-OC(O)R70、-OC(S)R70、-OC(O)O-M+、-OC(O)OR70、-OC(S)OR70、-NR70C(O)R70、-NR70C(S)R70、-NR70CO2–M+、-NR70CO2R70、-NR70C(S)OR70、-NR70C(O)NR80R80、-NR70C(NR70)R70和-NR70C(NR70)NR80R80,其中R60選自任選取代的烷基、環烷基、雜環烷基、雜環烷基烷基、環烷基烷基、芳基、芳基烷基、雜芳基和雜芳基烷基,各R70獨立地為氫或R60;各R80獨立地為R70,或者,兩個R80',與它們連接的氮原子一起形成3-、4-、5-、6-、或7-元雜環烷基,該雜環烷基可任選包含1至4個相同或不同的選自O、N和S的其它雜原子,其中N可具有–H、C1-C4烷基、-C(O)C1-4烷基、-CO2C1-4烷基、或–SO2C1-4烷基取代;且各M+為具有凈單一正電荷的抗衡離子。各M+可獨立地為,例如,堿金屬離子,如K+、Na+、Li+;銨離子,如+N(R60)4;或堿土金屬離子,如[Ca2+]0.5、[Mg2+]0.5或[Ba2+]0.5("下標0.5是指該二價堿土金屬離子的一個抗衡離子可為該實施方案的化合物的離子化形式,而另一典型抗衡離子如氯離子,或者本文公開的兩個離子化化合物可以用作這樣的二價堿土金屬離子的抗衡離子,或該實施方案的雙離子化化合物可用作這種二價堿土離子的抗衡離子)。
除了本文的公開內容,在"取代的"烯烴、炔烴、芳基和雜芳基中不飽和碳原子上氫的取代基,除非另有所述,為-R60、鹵素、-O-M+、-OR70、-SR70、-S–M+、-NR80R80、三鹵代甲基、-CF3、-CN、-OCN、-SCN、-NO、-NO2、-N3、-S(O)R70、-SO2R70、-SO3–M+、-SO3R70、-OSO2R70、-OSO3–M+、-OSO3R70、-PO3-2(M+)2、-P(O)(OR70)O–M+、-P(O)(OR70)2、-C(O)R70、-C(S)R70、-C(NR70)R70、-CO2–M+、-CO2R70、-C(S)OR70、-C(O)NR80R80、-C(NR70)NR80R80、-OC(O)R70、-OC(S)R70、-OCO2–M+、-OCO2R70、-OC(S)OR70、-NR70C(O)R70、-NR70C(S)R70、-NR70CO2–M+、-NR70CO2R70、-NR70C(S)OR70、-NR70C(O)NR80R80、-NR70C(NR70)R70和-NR70C(NR70)NR80R80,其中R60、R70、R80和M+如之前所定義,條件是在取代的烯烴或炔烴的情況下,所述取代基不為-O-M+、-OR70、-SR70或-S–M+。
除了本文關于單獨術語公開的取代基,在“取代的”雜環烷基和環烷基中氮原子上的氫的取代基,除非另有所述,為-R60、-O-M+、-OR70、-SR70、-S-M+、-NR80R80、三鹵代甲基、-CF3、-CN、-NO、-NO2、-S(O)R70、-S(O)2R70、-S(O)2O-M+、-S(O)2OR70、-OS(O)2R70、-OS(O)2O-M+、-OS(O)2OR70、-P(O)(O-)2(M+)2、-P(O)(OR70)O-M+、-P(O)(OR70)(OR70)、-C(O)R70、-C(S)R70、-C(NR70)R70、-C(O)OR70、-C(S)OR70、-C(O)NR80R80、-C(NR70)NR80R80、-OC(O)R70、-OC(S)R70、-OC(O)OR70、-OC(S)OR70、-NR70C(O)R70、-NR70C(S)R70、-NR70C(O)OR70、-NR70C(S)OR70、-NR70C(O)NR80R80、-NR70C(NR70)R70和-NR70C(NR70)NR80R80,其中R60、R70,R80和M+如之前所定義。
除了本文公開的內容,在某一實施方案中,經取代的基團具有1、2、3或4個取代基,1、2或3個取代基,1或2個取代基,或1個取代基。
應理解,在上文所定義的所有取代基團中,本文不欲將通過限定本身具有其他取代基的取代基而得到的聚合物(例如,具有取代的芳基作為取代基的取代的芳基,該取代基本身由取代的芳基取代,該取代的芳基進一步由取代的芳基取代,等)包括在內。在此類情況下,這些取代的最大數目為3。例如,本文特定涵蓋的取代的芳基的連續取代限于取代的芳基-(取代的芳基)-取代的芳基。
除非另有說明,否則本文未明確定義的取代基的命名法是通過命名官能團的末端部分然后命名朝向連接點的相鄰官能團而獲得。例如,取代基“芳烷氧羰基”是指基團(芳基)-(烷基)-O-C(O)-。
關于本文公開的含有一或多個取代基的任何基團,應理解,顯然,這些基團不含有在空間上不能實現和/或合成上不可行的任何取代或取代方式。另外,主題化合物包括由這些化合物的取代產生的所有立體化學異構體。
術語“藥物可接受的鹽”意指對于給藥至患者(例如哺乳動物)可接受的鹽(對給定劑量方案具有可接受的哺乳動物安全性的含抗衡離子的鹽)。此類鹽可以來源于藥物可接受的無機或有機堿,以及來源于藥物可接受的無機或有機酸。“藥物可接受的鹽”是指來源于本領域中熟知的各種有機及無機抗衡離子的化合物的藥物可接受的鹽,且僅舉例而言,包括鈉鹽、鉀鹽、鈣鹽、鎂鹽、銨鹽、四烷基銨鹽等;且當分子含有堿性官能團時,包括有機或無機酸的鹽,如鹽酸鹽、氫溴酸鹽、甲酸鹽、酒石酸鹽、苯磺酸鹽、甲磺酸鹽、乙酸鹽、馬來酸鹽、草酸鹽等。
基團結構圖中的波浪線代表基團至母體結構的連接點。
術語“其鹽”意指當酸的質子由陽離子(例如金屬陽離子或有機陽離子等)替換時所形成的化合物。在適用情況下,該鹽為藥學上可接受的鹽,但此條件對不預期給藥至患者的中間體化合物的鹽而言并非必需。舉例而言,本化合物的鹽包括其中通過無機或有機酸來質子化以形成陽離子的那些化合物,其中無機或有機酸的共軛堿作為鹽的陰離子組分。
“溶劑合物”是指通過溶劑分子與溶質的分子或離子的組合形成的復合物。溶劑可以是有機化合物,無機化合物或兩者的混合物。溶劑的一些實例包括但不限于甲醇,N,N-二甲基甲酰胺,四氫呋喃,二甲基亞砜和水。當溶劑是水時,形成的溶劑合物是水合物。
“立體異構體”是指具有相同原子連接性但在空間中具有不同原子排列的化合物。立體異構體包括順式-反式異構體、E和Z異構體、對映異構體和非對映異構體。
“互變異構體”是指僅在原子的電子連接和/或在質子位置方面不同的分子的替代形式,例如烯醇-酮和亞胺-烯胺互變異構體,或含有-N=C(H)-NH-環原子排列的雜芳基的互變異構形式,例如吡唑、咪唑、苯并咪唑、三唑和四唑。本領域普通技術人員將認識到其它互變異構的環原子排列是可能的。
應當理解,術語“或其鹽或溶劑合物或立體異構體”旨在包括鹽,溶劑合物和立體異構體的所有排列,例如主題化合物的立體異構體的藥學上可接受的鹽的溶劑合物。
如本文所用,藥物、化合物、綴合物、藥物綴合物、抗體藥物綴合物或藥物組合物的“有效劑量”或“有效量”是足以實現有益或所需結果的量。對于預防性使用,有益或期望的結果包括諸如消除或降低疾病的風險,減輕其嚴重性或延遲疾病發作的結果,包括在疾病的發展過程中疾病的生化、組織學和/或行為癥狀,其并發癥和中間病理表型。對于治療用途,有益或期望的結果包括臨床結果,例如減少由疾病導致的一種或多種癥狀,增加患有疾病的那些人的生活質量,減少治療疾病所需的其它藥物的劑量,例如通過靶向增強另一種藥物的效果、延遲疾病的進展,和/或延長存活。在癌癥或腫瘤的情況下,有效量的藥物可以具有以下效果:減少癌細胞數目;減少腫瘤大小;抑制(即,在一定程度上減緩并且優選停止)癌細胞浸潤到外周器官中;抑制(即,在一定程度上減緩并且優選停止)腫瘤轉移;在一定程度上抑制腫瘤生長;和/或在一定程度上緩解與疾病相關的一種或多種癥狀。有效劑量可以一次或多次施用來施用。出于本公開的目的,藥物、化合物或藥物組合物的有效劑量是足以直接或間接實現預防性或治療性治療的量。如在臨床背景中所理解的,藥物、化合物或藥物組合物的有效劑量可以或可以不與另一種藥物、化合物或藥物組合物聯合實現。因此,在給予一種或多種治療劑的上下文中可以考慮“有效劑量”,并且如果與一種或多種其他藥劑聯合,可以認為單一試劑以有效量給予,期望的結果可以被實現或被實現。
如本文所用,“給藥”是指用于分配治療劑以治療病癥的通用術語。在一些實施方案中,本公開的藥物組合物含有適于使化合物或混合物作為片劑、膠囊或丸劑口服給藥的藥學上可接受的載體或賦形劑,或使得化合物適于腸胃外、靜脈內、皮內、肌內、腹膜內、鼻內、舌下、氣管內、吸入、眼、陰道、直腸、皮下或經皮施用的藥學上可接受的載體或賦形劑。
有多種給藥途徑。所選擇的具體模式當然取決于所選擇的具體藥劑、所治療的具體病癥和治療功效所需的劑量。下面討論幾種給藥方式。
本發明的化合物或藥物組合物的給藥可通過本領域技術人員已知的任何方式實現。給藥途徑包括但不限于口服、腸胃外、靜脈內、肌內、腹腔內、鼻內、舌下、氣管內、吸入、皮下、眼、陰道和直腸的。全身途徑包括口服和腸胃外。
對于口服給藥,可以通過將活性化合物與本領域熟知的藥學上可接受的載體組合來容易地配制。這樣的載體使得本公開的化合物能夠配制為用于由待治療的受試者口服攝取的片劑、丸劑、糖錠劑、膠囊、液體、凝膠、糖漿、漿料、懸浮液等。任選地,口服制劑還可以配制在用于中和內部酸性條件的鹽水或緩沖液中,或者可以在沒有任何載體的情況下施用。
當需要將它們全身遞送時,化合物可以配制成用于通過注射的腸胃外給藥,例如通過推注(bolus injection)或連續輸注。用于注射的制劑可以以單位劑型存在,例如在安瓿或多劑量容器中,加入防腐劑。組合物可以采取在油性或水性載體中的懸浮液、溶液或乳液的形式,并且可以含有配制劑如懸浮劑、穩定劑和/或分散劑。
用于腸胃外給藥的藥物制劑包括水溶性形式的活性化合物的水溶液。此外,活性化合物的懸浮液可以制備為適當的油性注射懸浮液。合適的親脂性溶劑或載體包括脂肪油如芝麻油,或合成脂肪酸酯,如油酸乙酯或甘油三酯,或脂質體。水性注射懸浮液可以含有增加懸浮液粘度的物質,例如羧甲基纖維素鈉、山梨醇或葡聚糖。任選地,懸浮液還可以含有合適的穩定劑或增加化合物的溶解性以允許制備高度濃縮溶液的試劑。或者,活性化合物可以是粉末形式,在使用前用合適的載體例如無菌無熱原水重構。
化合物還可以配制成直腸或陰道組合物,例如栓劑或保留灌腸劑,例如含有常規栓劑基質如可可脂或其它甘油酯。
如本文所使用的,“與……聯合”是指除了一種治療方式之外施用另一種治療方式。因此,“與……聯合”是指在向個體施用其它治療方式之前、期間或之后施用一種治療方式。
如本文所用,“治療”是用于獲得有益或期望的結果的方法,包括并優選臨床結果。為了本公開的目的,有益或期望的臨床結果包括但不限于以下的一種或多種:減少(或破壞)癌細胞的增殖,減少由疾病導致的癥狀,增加患有疾病的人的生活質量,減少治療疾病所需的其它藥物的劑量,延遲疾病的進展和/或延長個體的存活。
如本文所用,“延遲疾病的發展”是指延遲、阻礙、減緩、延遲、穩定和/或推遲疾病(例如癌癥)的發展。這種延遲可以具有變化的時間長度,這取決于疾病和/或被治療的個體的病史。對于本領域技術人員明顯的是,足夠的或顯著的延遲實際上可包括預防,即個體不發展疾病。例如,晚期癌癥,例如轉移的發展可能被延遲。
“個體”或“受試者”是哺乳動物,更優選人。哺乳動物還包括但不限于農場動物、運動動物、寵物(例如貓、狗、馬)、靈長類動物、小鼠和大鼠。“在有需要的個體中治療癌癥”是被鑒定為患有癌癥的個體,即個體已經由醫生(例如使用本領域眾所周知的方法)診斷為患有癌癥。在一些實施方案中,需要治療的個體是懷疑患有或發展癌癥的個體。懷疑患有或發展癌癥的個體的實例包括但不限于被鑒定為具有與癌癥或癌癥發展相關的突變的受試者,具有癌癥家族史的受試者以及先前或曾經已經治愈癌癥的受試者(包括緩解期的癌癥患者)。
如本文所用,術語“特異性識別”或“特異性結合”是指可測量的和可再現的相互作用,例如靶標和抗體(或分子或部分)之間的吸引或結合,其在存在包括生物分子在內的分子的異質群體的情況下決定靶標的存在。例如,特異性或優先結合表位的抗體是以比其結合靶標或非靶標表位的其它表位更大的親和力、活動性、更容易和/或更長的持續時間結合該表位的抗體。還應理解,例如,特異性或優先結合第一靶標的抗體(或部分或表位)可以或可以不特異性或優先結合第二靶標。因此,“特異性結合”或“優先結合”不一定需要(盡管其可以包括)排他性結合。特異性結合靶標的抗體可具有至少約10 3M-1或10 4M-1,有時約10 5M-1或10 6M-1,在其它情況下約10 6M-1或10 7M-1,約10 8M-1至10 9M-1,或約10 10M-1至10 11M-1或更高的締合常數。多種免疫測定形式可用于選擇與特定蛋白質特異性免疫反應的抗體。例如,固相ELISA免疫測定常規用于選擇與蛋白質特異性免疫反應的單克隆抗體。關于可用于測定特異性免疫反應性的免疫測試形式和條件的描述,參見例如Harlow和Lane(1988)Antibodies,A Laboratory Manual,Cold Spring Harbor Publications,New York。
如本文所用,術語“癌癥”、“腫瘤”、“癌性”和“惡性”是指或描述哺乳動物中通常特征在于不受調節的細胞生長的生理狀況。癌癥的實例包括但不限于癌,包括腺癌、淋巴瘤、母細胞瘤、黑素瘤和肉瘤。這種癌癥的更具體的實例包括鱗狀上皮細胞癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺鱗狀細胞癌、胃腸癌、霍奇金淋巴瘤和非霍奇金淋巴瘤、胰腺癌、成膠質細胞瘤、子宮頸癌、神經膠質瘤、卵巢癌、肝癌(liver cancer)如肝臟癌瘤(hepatic carcinoma)和肝癌(hepatoma)、膀胱癌、乳腺癌、結腸癌、結腸直腸癌、子宮內膜或子宮癌、唾液腺癌、腎癌如腎細胞癌和維爾姆斯腫瘤、基底細胞癌、黑素瘤、間皮瘤、前列腺癌、甲狀腺癌、睪丸癌、食管癌、膽囊癌和各種類型的頭頸癌。
如本文和所附權利要求中所使用的,單數形式“一”、“一個”和“該”包括復數指代,除非上下文另有明確指示例如,提及“抗體”是指一至多個抗體,例如摩爾量,并且包括本領域技術人員已知的等同物,等等。
本文中提及“約”一個值或參數包括(并描述)涉及該值或參數本身的實施方案。例如,涉及“約X”的描述包括對“X”的描述。
應當理解,本文所述的本公開的方面和變化包括“由……組成”和/或“基本上由……組成”的方面和變化。
除非另有定義,本文使用的所有技術和科學術語具有與本公開所屬領域的普通技術人員通常理解的相同的含義。盡管與本文所述的那些類似或等同的任何方法和材料也可以用于本公開的實踐或測試,現在描述優選的方法和材料。本文提及的所有出版物通過引用并入本文,以公開和描述與所引用的出版物相關的方法和/或材料。
除非另有說明,本實施方案的方法和技術通常根據本領域公知的常規方法進行,并且在本說明書通篇引用和討論的各種一般和更具體的參考文獻中描述。參見例如Loudon,Organic Chemistry,第4版,New York:Oxford University Press,2002,第360-361、1084-1085頁;Smith和March,March's Advanced Organic Chemistry:Reactions,Mechanisms,and Structure,5th edition,Wiley-Interscience,2001。
本文使用的命名主題化合物的命名法在本文的實施例中說明。該命名通常使用可商購的AutoNom軟件(MDL,San Leandro,Calif.)得到。
應當理解,為了清楚起見,在單獨實施方案的背景中描述的本公開的某些特征也可以在單個實施方案中組合提供。相反,為了簡潔,在單個實施方案的背景中描述的本公開的各種特征也可以單獨地或以任何合適的亞組合提供。涉及由變量表示的化學基團的實施方案的所有組合特別地包括在本公開內容中并且在本文中公開,就好像各個和每個組合單獨和明確地公開,只要這樣的組合包括的化合物為穩定化合物(即,可以分離、表征和測試生物活性的化合物)。此外,描述這些變量的實施方案中列出的化學基團的所有亞組合也特別包括在本公開中,并且在本文中公開,就如同化學基團的各個和每個這樣的亞組合在本文中單獨和明確地公開。
本發明提供式(I)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
X為親水性自降解連接基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵或自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
本發明還提供式(II)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
在一些實施方案中,該靶向部分具有一個或多個用于連接至藥物部分的連接位點。例如,靶向部分T可具有多個位點以用于連接至連接基-藥物部分(例如,A-L4-L3-L2-X-L1-D)。因此,還提供了式(Ia)的化合物:
或其鹽或溶劑合物或立體異構體,其中X,L1,L2,L3,L4和A如式(I)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
本發明還提供式(IIa)的化合物:
或其鹽或溶劑合物或立體異構體,其中R1,X,L1,L2,L3,L4和A如式(II)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。
肽連接基
在一些實施方案中,L3為肽連接基。在一些實施方案中,L3為1、2、3、4、5、6、7、8、9或10個氨基酸殘基的肽連接基。在某些實施方案中,L3為2、3或4個氨基酸殘基的肽連接基。在一些情況下,L3為二肽連接基。
氨基酸殘基可為天然存在的或非天然氨基酸殘基。術語“天然氨基酸”和“天然存在的氨基酸”是指Ala、Asp、Cys、Glu、Phe、Gly、His、Ile、Lys、Leu、Met、Asn、Pro、Gln、Arg、Ser、Thr、Val、Trp和Tyr。“非天然氨基酸”(即,氨基酸不是天然存在)包括,作為非限制性的實例,高絲氨酸、高精氨酸、瓜氨酸、苯基甘氨酸、牛磺酸、碘化酪氨酸(iodotyrosine)、硒代半胱氨酸、正亮氨酸(“Nle”)、正纈氨酸(“Nva”)、β-丙氨酸、L-或D-萘基丙氨酸、鳥氨酸(“Orn”),等。
氨基酸還包括天然和非天然氨基酸的D-形式。“D-”表示具有“D”(右旋)構型的氨基酸,與天然存在(“L-”)氨基酸中的構型相反。在沒有指明具體構型的情況下,本領域技術人員將理解氨基酸是L-氨基酸。然而,氨基酸也可以是D-和L-構型的外消旋混合物。天然和非天然氨基酸可商購(Sigma Chemical Co.;Advanced Chemtech)或使用本領域已知的方法合成。可以基于殘基的極性,電荷,溶解性,疏水性,親水性和/或兩親性性質的相似性進行氨基酸替代,只要保留其生物活性即可。
氨基酸殘基序列可以被特異性修飾,使得其將通過一種或多種腫瘤相關蛋白酶從所得的肽基衍生物藥物-綴合物選擇性地酶斷裂。
在某些實施方案中,L3為包含至少一個賴氨酸或至少一個精氨酸殘基的肽連接基。
在某些實施方案中,L3為包含選自以下的氨基酸殘基的肽連接基:賴氨酸、D-賴氨酸、瓜氨酸、精氨酸、脯氨酸、組氨酸、鳥氨酸和谷氨酰胺。
在某些實施方案中,L3為包含選自以下的氨基酸殘基的肽連接基:纈氨酸、異亮氨酸、苯丙氨酸、蛋氨酸、天冬酰胺、脯氨酸、丙氨酸、亮氨酸、色氨酸和酪氨酸。
在某些實施方案中,L3為選自以下的二肽連接基:纈氨酸-瓜氨酸、脯氨酸-賴氨酸、蛋氨酸-D-賴氨酸、天冬酰胺-D-賴氨酸、異亮氨酸-脯氨酸、苯丙氨酸-賴氨酸和纈氨酸-賴氨酸。在某些實施方案中,L3為纈氨酸-瓜氨酸。
在其對特定腫瘤相關蛋白酶的酶斷裂的選擇性方面,可以設計和優化適于在本公開中使用的許多特異性肽連接基分子。用于本公開的某些肽連接基是針對蛋白酶、組織蛋白酶B和D優化的那些。
親水性自降解連接基
在本文所述的化合物的一些實施方案中,X為親水性自降解連接基。
本公開的化合物采用親水性自降解間隔基部分,其將兩個官能部分間隔和共價連接在一起,并引入親水基團,其提供化合物的更好的溶解性。在一些實施方案中,親水性自降解間隔基部分將靶向部分和藥物部分連接在一起。一些酶不穩定連接基的增加的相關疏水性可導致藥物綴合物的聚集,特別是對于強疏水性藥物。通過將親水基團引入連接基中,藥物綴合物的聚集將減少。
自降解間隔基可以定義為雙官能化學部分,其能夠將兩個間隔開的化學部分共價連接在一起成為通常穩定的三部分分子,可以通過酶斷裂從三部分分子中釋放一個間隔的化學部分;并且在酶斷裂后,可以自發地從分子的其余部分斷裂以釋放另一個間隔的化學部分。
在某些實施方案中,X為芐基氧基羰基。在某些實施方案中,X為
其中R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基。
在一些實施方案中,本發明提供式(II)的化合物:
或其鹽或溶劑合物或立體異構體;
其中:
D為藥物部分;
T為靶向部分;
R1為氫、未取代或取代的C1-3烷基、或未取代或取代的雜環基;
L1為鍵、自降解連接基或環化自消除型連接基;
L2為鍵、自降解連接基;
其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;
其中如果L2為自降解連接基,則L1為鍵;
L3為肽連接基;
L4為鍵或間隔基;且
A為酰基單元。
還提供了式(IIa)的化合物:
或其鹽或溶劑合物或立體異構體;其中D、T、L1、L2、L3、L4和A如式(II)所定義,且p為1至20。在一些實施方案中,p為1至8。在一些實施方案中,p為1至6。在一些實施方案中,p為1至4。在一些實施方案中,p為2至4。在一些實施方案中,p為1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些實施方案中,p為1、2、3或4。在一些實施方案中,p為2。在一些實施方案中,p為3。在一些實施方案中,p為4。
在式(II)或(IIa)的某些實施方案中,R1為氫。在一些情況下,R1為甲基。
藥物部分的釋放是基于氨基芐基氧基羰基的自消除反應。出于解釋目的,連接有藥物和肽的氨基芐基氧基羰基的反應方案示于以下。
方案1
參考方案1,在從肽斷裂后,形成氨基芐基氧基羰基且其能經歷自發的1,6消除以形成環己-2,5-二烯亞胺衍生物和二氧化碳且釋放藥物。
任選的第二自降解連接基或環化自消除型連接基
任選的第二自降解連接基或環化自消除型連接基提供另外的連接基以允許精細調整化合物的斷裂以釋放藥物部分。
在本文所述的化合物中,L1為鍵、自降解連接基或環化自消除型連接基;L2為鍵或自降解連接基;其中如果L1為自降解連接基或環化自消除型連接基,則L2為鍵;且其中如果L2為自降解連接基,則L1為鍵。因此,存在任選的第二自降解連接基或環化自消除型連接基與該親水性自降解連接基相鄰。
在某些實施方案中,L1為鍵且L2為鍵。在某些實施方案中,L1為自降解連接基或環化自消除型連接基且L2為鍵。在某些實施方案中,L1為鍵且L2為自降解連接基。
在一些實施方案中,L1為鍵。在某些實施方案中,L1為自降解間隔基或環化自消除型連接基,其將親水性自降解連接基和藥物部分分離。在某些實施方案中,L1為氨基芐基氧基羰基連接基。
在某些實施方案中,L1選自:
其中n為1或2。
在一些情況下,該自降解連接基或環化自消除型連接基為可使用的更多種的部分提供設計可能性。例如,在式(IV)或(IVa)中,親水性自降解連接基和藥物部分之間的氨基甲酸酯連接(-O-C(O)-N(H)-)將提供穩定的藥物綴合物且將易于斷裂以提供游離藥物部分。該親水性自降解連接基通常以氧基羰基(-O-C(O)-)結尾。如果該藥物部分具有可用于反應以形成氨基甲酸酯基的氨基-反應性基團,則該(任選的)第二自降解單元或環化自消除型連接基不是必需的;但其仍可使用。然而,如果該藥物不包含氨基,而是包含一些其它反應性官能團,則該藥物仍可通過在藥物部分和氨基芐基氧基羰基之間包含中間的自降解間隔基或環化自消除型連接基,而摻入本實施方案的包含氨基芐基氧基羰基的化合物中。
以下L1的環化自消除型連接基提供包含羥基的或包含巰基的藥物部分與親水性自降解連接基的氨基芐基氧基羰基的連接:
該實施方案的化合物中的環化自消除型連接基提供化合物的斷裂以釋放藥物部分。相鄰親水性自降解連接基的消除機理將釋放L1的氨基。該氨基然后可在環化反應中與L1的氨基甲酸酯基或硫代氨基甲酸酯連接和藥物部分反應以釋放含羥基或含巰基的藥物部分。
在一些實施方案中,L2為鍵。在某些實施方案中,L2為自降解間隔基,其將親水性自降解連接基與肽連接基分開。在某些實施方案中,L2為氨基芐基氧基羰基連接基。
在某些實施方案中,L2選自
其中n為1或2。
任選的間隔基
在一些實施方案中,L4為鍵或間隔基。在某些實施方案中,L4為鍵。在某些實施方案中,L4為間隔基,其可提供在藥物部分和靶向部分之間的距離。
在某些實施方案中,間隔基選自烷基、取代的烷基、烯基、取代的烯基、炔基、取代的炔基、芳基、取代的芳基、環烷基、取代的環烷基、雜芳基、取代的雜芳基、雜環基、取代的雜環基、和雜原子,及其組合。該間隔基在其原子組成中可為同質的或異質的(例如,僅包含碳原子的間隔基或在間隔基上存在碳原子和一種或多種雜原子的間隔基)。優選地,該間隔基包含1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、或50個碳原子和0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30個選自氧、氮和硫的雜原子。該間隔基也可為手性或非手性的,直鏈、支鏈的或環狀的。
在某些實施方案中,L4為選自以下的間隔基:聚亞烷基二醇、亞烷基、亞烯基、亞炔基和聚胺。亞烯基的實例包括,但不限于,亞乙烯基(-CH=CH-)、亞烯丙基(-CH2C=C-)和亞丁-3-烯-1-基(-CH2CH2C=CH-)。亞烯基的實例包括,但不限于,亞乙炔基(-C≡C-)和亞炔丙基(-CH2C≡C-)。
在某些實施方案中,L4為間隔基,其包含可提供與肽鍵末端的連接的官能團。官能團,如C(O)、C(O)-NH、S(O)2和S(O)2-NH,可提供與肽鍵末端的連接。在一些情況下,L4為L4a-C(O)、L4a-C(O)-NH、L4a-S(O)2、L4a-S(O)2-NH,其中L4a選自聚亞烷基二醇、亞烷基、亞烯基、亞炔基和聚胺。在一些情況下,L4為L4a-C(O),其中L4a選自聚亞烷基二醇、亞烷基、亞烯基、亞炔基和聚胺。
在某些實施方案中,L4為L4a-C(O),其中L4a為聚亞烷基二醇。在某些實施方案中,L4為L4a-C(O),其中L4a為聚乙二醇。在某些實施方案中,所述間隔基為式-CH2-(CH2-O-CH2)m-CH2-C(O)-,其中m為整數0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30。
在某些實施方案中,L4為L4a-C(O),其中L4a為亞烷基。在某些實施方案中,L4為L4a-C(O),其中L4a為C1-10亞烷基、C1-8亞烷基或C1-6亞烷基。在某些實施方案中,L4為L4a-C(O),其中L4a為C4亞烷基、C5亞烷基或C6亞烷基。在某些實施方案中,L4為L4a-C(O),其中L4a為C5亞烷基。
酰基單元
在本文所述的化合物中,A為酰基單元。在某些實施方案中,該酰基單元“A”包含硫原子且通過衍生自靶向部分的硫原子連接至靶向部分。在該情況下,二硫鍵在酰基單元和靶向部分之間形成。
在某些實施方案中,A選自
其中Q2為NH或O,各q獨立地為1至10的整數,且各q1獨立地為1至10的整數。在一些實施方案中,q為整數2至5,如2、3、4或5。在一些實施方案中,q1為整數2至5,如2、3、4或5。
在某些實施方案中,A為其中Q2為NH或O且q為整數1、2、3、4、5、6、7、8、9或10。在某個情況下,q為2至5的數,如2、3、4或5。
在某些實施方案中,A為其中Q2為NH或O且q為整數1、2、3、4、5、6、7、8、9或10。在某個情況下,q為2至5的數,如2、3、4或5。
在某些實施方案中,A選自
其中Q2為NH或O。
藥物部分
本實施方案的藥物綴合物對于通常目的是有效的(其中相應藥物是有效的),并有卓越的功效,因為其在靶向部分所固有的將藥物輸送到期望的細胞(在該細胞中,所述藥物是特別有益的)的能力。
用于本發明實施方案的優選藥物是細胞毒性藥物,例如用于癌癥治療的那些。這樣的藥物通常包括DNA損傷劑、抗代謝物、天然產物及其類似物。某些類型的細胞毒性劑包括例如酶抑制劑,例如二氫葉酸還原酶抑制劑、胸苷酸合酶抑制劑、DNA嵌入劑、DNA斷裂劑、拓撲異構酶抑制劑、蒽環類家族藥物、長春花藥物、絲裂霉素、博萊霉素、細胞毒性核苷、蝶啶家族藥物、二炔類、鬼臼毒素、分化誘導劑和紫杉醇。這些類別的某些有用成員包括例如甲氨蝶呤、甲基葉酸、二氯甲氨蝶呤、5-氟尿嘧啶、6-巰基嘌呤、阿糖胞苷、美法侖、長春羅新、白諾西丁(leurosideine)、放線菌素、柔紅霉素、多柔比星、絲裂霉素C、絲裂霉素A、洋紅霉素、氨基蝶呤、他利霉素、鬼臼毒素和鬼臼毒素衍生物如依托泊苷或磷酸依托泊苷、長春堿、長春新堿、長春地辛、紫杉醇、泰索帝、視黃酸、丁酸、N8-乙酰基亞精胺、喜樹堿、以及它們的類似物。其它藥物包括多拉司他汀和多卡霉素。
本領域技術人員可對所需化合物進行化學修飾,以使該化合物的反應對于制備本公開的綴合物更為方便。
在某些實施方案中,D為具有化學反應性官能團的藥物部分,通過該化學反應性官能團該藥物連接至L1或X。在一些情況下,該官能團選自伯胺、仲胺、羥基和巰基。在一些情況下,該官能團為伯胺或仲胺。在一些情況下,該官能團為羥基。在一些情況下,該官能團為巰基。
如上所述,該親水性自降解連接基通常以氧基羰基(-O-C(O)-)終止。因此,含氨基的藥物部分將易于與氧基羰基反應以形成氨基甲酸酯基。在某些實施方案中,D為含氨基的藥物部分,其中該藥物通過氨基連接至L1或X。
然而,如果該藥物部分不包含氨基,則L1的第二自降解連接基或環化自消除型連接基可為更多種可使用的部分提供設計可能性。在某些實施方案中,D為含羥基或含巰基的藥物部分,其中該藥物通過羥基或巰基連接至L1。
代表性含氨基的藥物包括絲裂霉素-C、絲裂霉素-A、柔紅霉素、多柔比星、氨基蝶呤、放線菌素、博來霉素、9-氨基喜樹堿、N8-乙酰基亞精胺、1-(2-氯乙基)-1,2-二甲烷磺酰基酰肼、他利霉素、阿糖胞苷、多拉司他汀以及它們的衍生物。含氨基的藥物還包括天然不包含氨基的藥物的氨基衍生物。在某些實施方案中,D為多卡霉素、多拉司他汀、tubulysin、多柔比星(DOX)、紫杉醇或絲裂霉素C(MMC),或其氨基衍生物。
代表性含羥基的藥物包括依托泊苷,喜樹堿、紫杉醇、埃斯培拉霉素、1,8-二羥基-雙環[7.3.1]十三碳-4-9-二烯-2,6-二炔-13-酮(美國專利5,198,560)、鬼臼毒素、anguidine、長春新堿、長春堿、嗎啉-多柔比星、正-(5,5-二乙酰氧基-戊基)多柔比星、多卡霉素,和它們的衍生物。
代表性含巰基的藥物包括埃斯培拉霉素和6-疏基嘌呤,和它們的衍生物。
用作本實施方案中的藥物的一組的細胞毒素劑包括下式的藥物:
(多拉司他汀的氨基衍生物)
(多卡霉素的氨基衍生物)
(多卡霉素的氨基衍生物)。
靶向部分
如本公開中所述的靶向部分是指與給定細胞群體特異性結合、復合、反應或締合的部分或分子。例如,靶向部分可以和與給定細胞群體(例如,尋求治療性處理或以其它方式生物修飾的給定細胞群體)相關的接受部分或受體特異性結合、復合、反應或締合。在本文所述的綴合物中,本文所述的靶向部分通過連接基與綴合物中的藥物部分連接。在一些實施方案中,靶向部分能夠將藥物部分(例如,用于治療目的的藥物部分)遞送至靶向部分與其結合、復合、反應或締合的特定靶細胞群。
該靶向部分可包括,例如,大分子量蛋白質,例如,抗體、較小分子量蛋白質、多肽或肽、和非肽基部分。本文所述的蛋白質、多肽或肽部分可包括,例如,轉鐵蛋白、血清白蛋白、表皮生長因子("EGF")、鈴蟾肽(bombesin)、胃泌素、胃泌素-釋放肽、血小板-衍生的生長因子、IL-2、IL-6、腫瘤生長因子("TGF")如TGF-α和TGF-β、痘苗病毒生長因子("VGF")、胰島素和胰島素樣生長因子I和II。非肽基部分可包括,例如,碳水化合物、凝集素和源自低密度脂蛋白的載脂蛋白。在某些實施方案中,蛋白質、抗體、多肽或肽可以指其未修飾的形式、已被修飾以用于本文所述的綴合物中的形式,例如用于與連接基結合的形式,或者在本文所述的綴合物中的部分。
在一些實施方案中,該靶向部分為抗體(或抗體部分或抗體靶向部分)。在一些實施方案中,該靶向部分包括抗體。在一些實施方案中,該靶向部分包括巰基(-SH)基團(例如,游離反應性巰基(-SH)基團)或可被修飾以包含該巰基。在一些實施方案中,該靶向部分包括具有巰基(例如,游離反應性巰基)的抗體。在一些實施方案中,該靶向部分包括游離巰基如具有游離巰基的抗體或可被修飾以包含該巰基。在一些實施方案中,包含巰基或硫醇基的該靶向部分通過巰基中的硫原子連接至連接基。
在一些實施方案中,該靶向部分(例如,抗體靶向部分)具有一個或多個連接位點以用于連接至藥物部分。例如,靶向部分T(例如,抗體)可具有多個位點(例如,多個巰基)以用于連接至連接基-藥物部分(例如,A-L4-L3-L2-X-L1-D,其中A適合于連接至靶向抗體的巰基)。在一些實施方案中,該靶向部分可具有1至20個連接位點。在一些實施方案中,該靶向部分可具有1至20,1至10,1至8,1至6,1至4,2至8,2至6,或2至4個連接位點。在一些實施方案中,該靶向部分具有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20個連接位點。在一些實施方案中,該靶向部分具有1,2,3,4,5,6,7,或8個連接位點。在一些實施方案中,該靶向部分具有2個連接位點。在一些實施方案中,該靶向部分具有1個連接位點。在一些實施方案中,該靶向部分具有4個連接位點。在一些情況下,某些潛在的連接位點可能不適于連接至藥物部分。因此,靶向部分T中的連接位點的數目可導致藥物綴合物具有比潛在的連接位點的數目更少數量的連接藥物部分。在一些實施方案中,一個或多個連接位點可以用于結合藥物部分。例如,抗體靶向部分可以在抗體的每條鏈上具有一個或兩個巰基,其適合通過連接基與藥物部分結合。
在一些實施方案中,該靶向部分是抗體或抗體靶向部分。本文所述的抗體是指能夠通過位于免疫球蛋白分子的可變區中的至少一個抗原識別位點特異性結合靶標(例如碳水化合物、多核苷酸、脂質、多肽等)的免疫球蛋白分子。如本文所用,術語“抗體”不僅包括完整的多克隆或單克隆抗體,而且包括其抗原結合片段(例如Fab、Fab’、F(ab’)2、Fv)、單鏈(ScFv)、其突變體、包含抗體部分的融合蛋白、以及包含抗原識別位點的免疫球蛋白分子的任何其它修飾的構型。抗體包括任何類別的抗體,例如IgG、IgA或IgM(或其亞類),并且抗體不需要是任何特定類別。根據其重鏈恒定結構域的抗體氨基酸序列,免疫球蛋白可以歸于不同類別。有五種主要類型的免疫球蛋白:IgA、IgD、IgE、IgG和IgM,其中幾種可以進一步分為亞類(同種型),例如IgG1、IgG2、IgG3、IgG4、IgA1和IgA2。對應于不同類別的免疫球蛋白的重鏈恒定結構域分別稱為α、δ、ε、γ和μ。不同類別的免疫球蛋白的亞基結構和三維構型是公知的。
包括或用于本文所述的靶向部分(或抗體靶向部分)中的抗體可包括單克隆抗體、多克隆抗體、抗體片段(例如Fab、Fab’、F(ab’)2、Fv、Fc等)、嵌合抗體、人源化抗體、人抗體(例如完全人抗體)、單鏈(ScFv)、雙特異性抗體、多特異性抗體、其突變體、包含抗體部分的融合蛋白、以及包含所需特異性的抗原識別位點的免疫球蛋白分子的任何其他修飾的構型。抗體可以是鼠、大鼠、駱駝、人或任何其它來源(包括人源化抗體)。在一些實施方案中,用于本文所述的靶向部分(或抗體靶向部分)中的抗體是以下任一種:對多肽具有親和力的雙特異性抗體、多特異性、單鏈、雙功能和嵌合和人源化分子,該親和力由所述抗體的至少一個高變區(HVR)或互補決定區(CDR)賦予。本公開中使用的抗體還包括單結構域抗體,其是抗體重鏈的可變結構域或抗體輕鏈的可變結構域。Holt等人,Trends Biotechnol.21:484-490,2003。現有技術也已知制備包含抗體重鏈的可變結構域或抗體輕鏈的可變結構域的域抗體的方法,其包含6種天然存在的來自抗體的HVR或CDR中的3種。參見,例如,Muyldermans,Rev.Mol.Biotechnol.74:277-302,2001。
在一些實施方案中,包括或用于本文所述的靶向部分(或抗體靶向部分)的抗體是單克隆抗體。如本文所用,單克隆抗體是指基本上同質性抗體的抗體,即構成群體的單個抗體是相同的,除了可能以少量存在的可能的天然存在的突變。此外,與通常包含針對不同決定簇(表位)的不同抗體的多克隆抗體制劑相反,單克隆抗體不是離散抗體的混合物。修飾語“單克隆”表示抗體從基本上同質的抗體群獲得的特征,并且不應解釋為需要通過任何特定方法產生抗體。例如,本公開中使用的單克隆抗體可以通過由Kohler和Milstein,1975,Nature,256:495首先描述的雜交瘤方法制備,或者可以通過重組DNA方法制備,如美國專利4,816,567所述。單克隆抗體還可以從例如使用McCafferty等人,1990,Nature,348:552-554中描述的技術產生的噬菌體庫中分離。
在一些實施方案中,包括或用于本文所述的靶向部分(或抗體靶向部分)的抗體是嵌合抗體。如本文所用,嵌合抗體指具有來自第一物種的可變區或可變區的部分和來自第二物種的恒定區的抗體。完整的嵌合抗體包含兩個拷貝的嵌合輕鏈和兩個拷貝的嵌合重鏈。嵌合抗體的產生是本領域已知的(Cabilly等人(1984),Proc.Natl.Acad.Sci.USA,81:3273-3277;Harlow和Lane(1988),Antibodies:a Laboratory Manual,Cold Spring Harbor Laboratory)。通常,在這些嵌合抗體中,輕鏈和重鏈的可變區都模擬來源于一種哺乳動物的抗體的可變區,而恒定部分與來源于另一種的抗體的序列同源。這種嵌合形式的一個明顯的優點是,例如,可變區可以方便地從目前已知的來源使用容易獲得的雜交瘤或來自非人宿主生物的B細胞與來源于例如人細胞制備物的恒定區組合而衍生。盡管可變區具有易于制備的優點,并且特異性不受其來源的影響,但是相比來自非人源的恒定區,當注射抗體時,作為人的恒定區不太可能引起來自人類受試者的免疫應答。然而,該定義不限于該特定示例。
在一些實施方案中,包括或用于本文所述的靶向部分(或抗體靶向部分)的抗體是人源化抗體。如本文所用,人源化抗體是指特異性嵌合免疫球蛋白、免疫球蛋白鏈或其片段(例如Fv、Fab、Fab'、F(ab')2或抗體的其它抗原結合亞序列)的非人(例如鼠)抗體,其含有源自非人免疫球蛋白的最小序列。大多數情況下,人源化抗體是人免疫球蛋白(受體抗體),其中來自受體的HVR或CDR的殘基被來自非人物種(供體抗體)的HVR或CDR的殘基替換,該非人物種例如具有所需特異性、親和力和能力的小鼠,大鼠,或兔。在一些情況下,人免疫球蛋白的Fv構架區(FR)殘基被相應的非人殘基替換。此外,人源化抗體可以包含既不在受體抗體中也不在輸入的HVR或CDR或框架序列中發現,但被包括以進一步改進和優化抗體性能的殘基。通常,人源化抗體將包含至少一個、通常兩個可變結構域的基本上全部,其中所有或基本上所有HVR或CDR區對應于非人免疫球蛋白的那些,并且全部或基本上全部FR區是人免疫球蛋白共有序列的FR區。人源化抗體最佳地還將包含免疫球蛋白恒定區或結構域(Fc)的至少一部分,通常是人免疫球蛋白的恒定區或結構域。抗體可具有如WO 99/58572中所述修飾的Fc區。其他形式的人源化抗體具有相對于原始抗體改變的一個或多個HVR或CDR(一個,兩個,三個,四個,五個,六個),其也稱為“衍生自”一個或更多個來自原始抗體的HVR或CDR的一個或多個HVR或CDR。
在一些實施方案中,包括或用于本文所述的靶向部分(或抗體靶向部分)的抗體是人抗體。如本文所用,人抗體指具有對應于由人產生的抗體的氨基酸序列的抗體,和/或已使用本領域已知的用于制備人抗體的任何技術制備的抗體。本文使用的人抗體包括包含至少一個人重鏈多肽或至少一個人輕鏈多肽的抗體。一個這樣的實例是包含鼠輕鏈和人重鏈多肽的抗體。人抗體可以使用本領域已知的多種技術產生。在一個實施方案中,人抗體選自噬菌體庫,其中噬菌體庫表達人抗體(Vaughan等人,1996,Nature Biotechnology,14:309-314;Sheets等人,1998,PNAS,95:6157-6162;Hoogenboom和Winter,1991,J.Mol.Biol.,227:381;Marks等人,1991,J.Mol.Biol.,222:581)。人抗體也可以通過將人免疫球蛋白基因座導入轉基因動物(例如其中內源免疫球蛋白基因已部分或完全失活的小鼠)來制備。該方法描述于美國專利5,545,807;5,545,806;5,569,825;5,625,126;5,633,425;和5,661,016。或者,人抗體可以通過使產生針對靶標抗原的抗體的人B淋巴細胞永生化來制備(所述B淋巴細胞可以從個體中回收或可以在體外免疫)。參見,例如,Cole等人,Monoclonal Antibodies and Cancer Therapy,Alan R.Liss,p.77(1985);Boerner等,1991,J.Immunol.,147(1):86-95;和美國專利號5,750,373。
人表皮生長因子2蛋白,HER2(ErbB2)受體酪氨酸激酶,已知在發育和腫瘤發生中都發揮關鍵作用。在一些實施方案中,包括或用于本文所述的靶向部分(或抗體靶向部分)的抗體特異性結合HER2。在進一步的實施方案中,抗HER2抗體是單克隆或人源化抗體。人源化單克隆抗HER2抗體曲妥珠單抗目前用于治療HER2-陽性癌癥。在一些實施方案中,包括在或用于本文所述的靶向部分中的抗體是曲妥珠單抗。其它抗HER2抗體,包括帕妥珠單抗和margetuximab,也是本領域已知的。在一些實施方案中,包括在或用于本文所述的靶向部分中的抗體是帕妥珠單抗。在一些實施方案中,包括在或用于本文所述的靶向部分中的抗體是margetuximab。
在一些實施方案中,該抗HER2抗體包含輕鏈可變區和/或重鏈可變區,該含輕鏈可變區包含1、2或3個源自SEQ ID NO:7的HVR(或CDR),該重鏈可變區包含1、2或3個源自SEQ ID NO:8的HVR(或CDR)。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含所述3個源自SEQ ID NO:7的HVR(或CDR),該重鏈可變區包含所述3個源自SEQ ID NO:8的HVR(或CDR)。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含的氨基酸序列至少約85%、至少約86%、至少約87%、至少約88%、至少約89%、至少約90%、至少約91%、至少約92%、至少約93%、至少約94%、至少約95%、至少約96%、至少約97%、至少約98%、或至少約99%與SEQ ID NO:7的序列相同,該重鏈可變區包含的氨基酸序列至少約85%、至少約86%、至少約87%、至少約88%、至少約89%、至少約90%、至少約91%、至少約92%、至少約93%、至少約94%、至少約95%、至少約96%、至少約97%、至少約98%、或至少約99%與SEQ ID NO:8的序列相同。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含SEQ ID NO:7的氨基酸序列,該重鏈可變區包含SEQ ID NO:8的氨基酸序列。
在一些實施方案中,該抗HER2抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含1、2或3個源自SEQ ID NO:12的HVR(或CDR),該重鏈可變區包含1、2或3個源自SEQ ID NO:13的HVR(或CDR)。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含所述3個源自SEQ ID NO:12的HVR(或CDR),該重鏈可變區包含所述3個源自SEQ ID NO:13的HVR(或CDR)。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含的氨基酸序列至少約85%,至少約86%,至少約87%,至少約88%,至少約89%,至少約90%,至少約91%,至少約92%,至少約93%,至少約94%,至少約95%,至少約96%,至少約97%,至少約98%,或至少約99%與SEQ ID NO:12的序列相同,該重鏈可變區包含的氨基酸序列至少約85%,至少約86%,至少約87%,至少約88%,至少約89%,至少約90%,至少約91%,至少約92%,至少約93%,至少約94%,至少約95%,至少約96%,至少約97%,至少約98%,或至少約99%與SEQ ID NO:13的序列相同。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含SEQ ID NO:12的氨基酸序列,該重鏈可變區包含SEQ ID NO:13的氨基酸序列。
在一些實施方案中,該抗HER2抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含1、2或3個源自SEQ ID NO:14的HVR(或CDR),該重鏈可變區包含1、2或3個源自SEQ ID NO:15的HVR(或CDR)。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含所述3個源自SEQ ID NO:14的HVR(或CDR),該重鏈可變區包含所述3個源自SEQ ID NO:15的HVR(或CDR)。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含的氨基酸序列至少約85%,至少約86%,至少約87%,至少約88%,至少約89%,至少約90%,至少約91%,至少約92%,至少約93%,至少約94%,至少約95%,至少約96%,至少約97%,至少約98%,或至少約99%與SEQ ID NO:14的序列相同,該重鏈可變區包含的氨基酸序列至少約85%,至少約86%,至少約87%,至少約88%,至少約89%,至少約90%,至少約91%,至少約92%,至少約93%,至少約94%,至少約95%,至少約96%,至少約97%,至少約98%,或至少約99%與SEQ ID NO:15的序列相同。在一些實施方案中,該抗體包含輕鏈可變區和/或重鏈可變區,該輕鏈可變區包含SEQ ID NO:14的氨基酸序列,該重鏈可變區包含SEQ ID NO:15的氨基酸序列。
表1:抗HER2ADC的序列ID NO.
人κ輕鏈恒定域序列(SEQ ID NO.1)RTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
人IgG1重鏈恒定域序列(SEQ ID NO.2)
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
人IgG2重鏈恒定域序列(SEQ ID NO.3)
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPSK
人IgG3重鏈恒定域序列(SEQ ID NO.4)
ASTKGPSVFPLAPCSRSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYTCNVNHKPSNTKVDKRVELKTPLGDTTHTCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFKWYVDGVEVHNAKTKPREEQYNSTFRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESSGQPENNYNTTPPMLDSDGSFFLYSKLTVDKSRWQQGNIFSCSVMHEALHNRFTQKSLSLSPGK
人IgG4重鏈恒定域序列(SEQ ID NO.5)
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPSCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
hIgG4-S228P的氨基酸序列(SEQ ID NO.6)
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
曲妥珠單抗(Roche Inc.)輕鏈可變區的氨基酸序列(SEQ ID NO.7)
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIK
曲妥珠單抗(Roche Inc.)重鏈可變區的氨基酸序列(SEQ ID NO.8)
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSS
包含人κ恒定域的曲妥珠單抗(Roche Inc.)輕鏈的氨基酸序列(SEQ ID NO.9)
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
包含人IgG1恒定域的曲妥珠單抗(Roche Inc.)重鏈的氨基酸序列(SEQ ID NO.10)
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
包含人IgG4-S228P恒定域的曲妥珠單抗(Roche Inc.)重鏈的氨基酸序列(SEQ ID NO.11)
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
帕妥珠單抗(Roche Inc.)輕鏈可變區的氨基酸序列(SEQ ID NO.12)
DIQMTQSPSSLSASVGDRVTITCKASQDVSIGVAWYQQKPGKAPKLLIYSASYRYTGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYIYPYTFGQGTKVEIK
帕妥珠單抗(Roche Inc.)重鏈可變區的氨基酸序列(SEQ ID NO.13)
EVQLVESGGGLVQPGGSLRLSCAASGFTFTDYTMDWVRQAPGKGLEWVADVNPNSGGSIYNQRFKGRFTLSVDRSKNTLYLQMNSLRAEDTAVYYCARNLGPSFYFDYWGQGTLVTVSS
Margetuximab(Macrogenics Inc.)輕鏈可變區的氨基酸序列(SEQ ID NO.14)
DIVMTQSHKFMSTSVGDRVSITCKASQDVNTAVAWYQQKPGHSPKLLIYSASFRYTGVPDRFTGSRSGTDFTFTISSVQAEDLAVYYCQQHYTTPPTFGGGTKVEIK
Margetuximab(Macrogenics Inc.)重鏈可變區的氨基酸序列(SEQ ID NO.15)
QVQLQQSGPELVKPGASLKLSCTASGFNIKDTYIHWVKQRPEQGLEWIGRIYPTNGYTRYDPKFQDKATITADTSSNTAYLQVSRLTSEDTAVYYCSRWGGDGFYAMDYWGQGASVTVSS
表2:抗HER2抗體曲妥珠單抗、帕妥珠單抗和Margetuximab的CDR的氨基酸序列
在一些實施方案中,包括在或用于本文所述的靶向部分中的抗體被修飾。在一些實施方案中,所述修飾為工程化的半胱氨酸取代。
在一些實施方案中,工程化的半胱氨酸取代發生在抗體的IgG重鏈上。在一些實施方案中,工程化的半胱氨酸取代發生在抗體的IgG重鏈上的特定位置。在一些實施方案中,可以具有工程化的半胱氨酸取代的IgG重鏈上的氨基酸位置包括(EU編號)118-215、234、235、236、237、238、239、246、248、249、254、265、267、269、270、273、276、278、279、282、283、284、286、287、289、292、293、294、297、298、299、300、302、303、312、314、315、318、320、324、326、327、330、332、333、334、335、336、337、339、341-447。上述公開的氨基酸位置描述于US 2012/0148580 A1;WO 2013/093809 A1;US 2009/0258420 A1;US 7521541 B2;US 7855275 B2;US 2011/0137017 A1;US 2012/0213705 A1;US 2011/0033378 A1;US 8455622 B2,其全部內容通過引用并入本文。可以是用于位點特異性綴合的工程化的半胱氨酸的IgG重鏈上的其它位置包括(EU編號)121、122、124、125、126、129、159、187、188、190、191、193、197、199、201、202、203、205、207、208、209、211、212、215、295、296、301。
在一些實施方案中,工程化的半胱氨酸取代發生在抗體的IgG輕鏈上。在一些實施方案中,工程化的半胱氨酸取代發生在抗體的IgG輕鏈上的特定位置。在一些實施方案中,可具有工程化的半胱氨酸取代的IgG輕鏈上的氨基酸位置包括(Kabat編號)108-211,如WO 2013/093809 A1;US 2009/0258420 A1;US 7855275B2;US 8455622B2中所述,其全部內容通過引用并入本文。可以是用于位點特異性綴合的工程化的半胱氨酸的IgG輕鏈上的另外的位置包括(Kabat編號)112、114、115、116、147、195、199、200、201、202、203、206、207、208、209、210。
治療應用
在一些實施方案中,本公開提供了通過向細胞施用足夠致死量的本文所討論的化合物來殺死細胞的方法。在一些實施方案中,被殺死的細胞是癌細胞。在一些實施方案中,所述細胞是乳腺癌細胞,或胃癌細胞或卵巢癌細胞。在一些實施方案中,本發明公開的殺死細胞的方法可以在體外進行。在一些實施方案中,殺死細胞的方法可以在體內進行。
在一些實施方案中,本文所討論的化合物可以作為治療受試者中疾病或病癥的治療方案的一部分以有效劑量施用。在一些實施方案中,本公開提供了用于治療有需要的個體中的癌癥的方法,其包括向個體施用有效劑量的本文所公開的化合物。在一些實施方案中,有效劑量為約0.001mg/kg至約1000mg/kg,約0.01mg/kg至約750mg/kg,約0.1mg/kg至約500mg/kg,約1.0mg/kg至約250mg/kg,約10.0mg/kg至約150mg/kg,一次或多次給藥,持續一天或幾天或多天,這取決于給藥方式和上述因素。
在一些實施方案中,本公開的化合物可以與其它治療化合物組合施用。在一些實施方案中,其它治療化合物是抗癌藥物或化學治療劑。本領域普通技術人員將熟悉多種癌癥化療劑。在一些實施方案中,本公開的化合物可以與其它形式的癌癥治療例如(但不限于)放射治療組合施用。
在一些實施方案中,本公開的治療癌癥的方法的使用導致有益的或期望的臨床結果,包括但不限于減少(或破壞)癌細胞的增殖,減少由疾病導致的癥狀,增加那些患有疾病的人的生活質量,和/或延緩疾病的發展。對于本領域技術人員明顯的是,足夠的或顯著的延遲實際上可包括預防,即該個體不產生癌癥。例如,晚期癌癥,例如轉移的發展可能被延遲。
在一些實施方案中,本文所述的化合物的靶向部分特異性結合癌細胞。癌癥結合靶向部分的示例性但非限制性實例包括抗CD20抗體,抗CD30抗體和抗HER2抗體。在進一步的實施方案中,本文所討論的化合物的藥物部分是有效治療癌癥的藥物。這種藥物的非限制性實例包括絲裂霉素-C、絲裂霉素-A、柔紅霉素、多柔比星、氨基蝶呤、放線菌素、博來霉素、9-氨基喜樹堿、N8-乙酰基亞精胺、1-(2-氯乙基)-1,2-二甲烷磺酰基酰肼、他利霉素、阿糖胞苷、多拉司他汀以及它們的衍生物。
提供以下實施例以說明但不限制本公開的效用。
實施例1
實施例2的材料和方法
連接基-藥物的合成
化合物Tap-18H的合成示于以下方案。中間體化合物M和O的合成也示于以下方案。
化合物TAP-18H的合成
化合物M的合成
化合物O的合成
參考化合物Tap-18H的合成方案,可商購的4-硝基苯基乙醛酸與N-甲基哌嗪縮合,其使用PCl5,或DMF中的EDCI和NiPr2Et,或CH2Cl2中的2-氯-4,6-二甲氧基-1,3,5-三嗪,且N-甲基嗎啉作為偶聯劑,以生成所需的酮酰胺。在典型的步驟中,在2-氯-4,6-二甲氧基-1,3,5-三嗪(5mmol)在CH2Cl2(20ml)中的溶液中,在0–5℃連續攪拌下添加N-甲基嗎啉(15mmol)。30–40分鐘后形成白色懸浮液,且向該混合物添加CH2Cl2(10ml)中的4-硝基苯基乙醛酸,導致形成澄清溶液。攪拌該混合物1小時后,在室溫添加N-甲基哌嗪(5mmol)。反應完成后(TLC,10分鐘),將混合物用10%NaHCO3水溶液(2×10ml)洗滌,然后用H2O(3×10ml)洗滌。有機層用無水硫酸鈉干燥且在減壓下去除溶劑得到粗產物,其進一步通過重結晶或柱色譜法(石油醚:乙酸乙酯=8:2)純化。
酮酰胺化合物進一步在THF或DIBAL-H或硼氫化鈉的存在下通過0.5當量的LiAlH4還原以產生硝基化合物C。[B.P.Bandgar和S.S.Pandit,Tetrahedron Letters 44(2003)3855–3858]
硝基化合物C通過用SnCl2處理或用Pd/C(10%w/w)作為催化劑在甲醇中在室溫下催化氫化約6-11小時而還原為苯胺化合物I,產率為65-81%。
它可以通過使用具有RB04-50反應器B的MultiMaxIR系統通過以下程序獲得。反應器最初用35ml甲醇、0.03mg 10%Pd/C和0.0252mol硝基化合物C填充,且在反應器中添加氫氣達到6.3巴(H2,恒定)的壓力。
參考合成化合物M的方案,將Boc-保護的L-纈氨酸用在DCM中的N-羥基琥珀酰亞胺和EDAC-HCl或在DCM中的N-羥基琥珀酰亞胺和EDC處理,得到琥珀酰亞胺酯。使該活化的酯與L-瓜氨酸和CH3CN、H2O、NaHCO3反應,得到Boc-保護的化合物M。
參考化合物Tap-18H的合成方案,通過DCC/HOBt在DMF中在室溫下將苯胺化合物I與Boc-保護的化合物M偶聯32小時,得到化合物N(產率78-82%),或與PS-碳二亞胺反應偶聯,在該反應中,在兩當量的PS-碳二亞胺和1.7當量的DCM中的HOBt的存在下,從100mg的化合物M與1.5當量的苯胺化合物I開始進行化合物N的合成,持續24小時。通過LC/MS分析顯示具有所需質量和約50-60%轉化率的峰。
偶聯的產物化合物N然后與4-硝基氯甲酸苯基酯在DCM中的2,6-二甲基吡啶的存在下在室溫反應8小時,以得到碳酸酯化合物P,LC/MS顯示具有所需質量的峰。
在DMF中的HOAt和Et3N的存在下用單甲基多拉司他汀10處理碳酸酯化合物P,導致形成化合物Q。
參考化合物O的合成方案,β-丙氨酸用馬來酸酐在DMF中處理,且如此得到的酸與N-羥基琥珀酰亞胺(NHS)在DCC偶聯下反應,以得到NHS-酯。可商購的t-boc-N-酰胺基-dPEG4-酸中的BOC保護基通過用TFA處理而去除以得到所述胺的TFA鹽,將其與之前合成的NHS酯反應。分離如此得到的羧酸且使用EDCI與N-羥基琥珀酰亞胺偶聯以得到NHS酯化合物O。
參考化合物Tap-18H的合成方案,化合物Q中的Boc-基團用TFA去除,且游離胺在無水乙腈和NaHCO3中在室溫與NHS酯化合物O偶聯12-36小時,以制備最終產物Tap-18H,產率為35-45%。
圖1顯示Tap-18H的NMR譜。
化合物TAP-18Hr1的合成
Tap-18Hr1通過下式合成。圖2顯示Tap-18Hr1的NMR譜。
化合物TAP-18Hr2的合成
Tap-18Hr2通過下式合成。圖3顯示Tap-18Hr2的NMR譜。
細胞系
人卵巢癌細胞SKOV-3(ATCC,目錄號HTB-77)在補充有10%FBS(HyClone,目錄號SH30071.03)、100U/mL青霉素/100μg/mL鏈霉素(GIBCO,目錄號15140)的McCoy's 5A培養基(修飾的)(GIBCO,目錄號16600)中培養。人乳腺癌細胞MDA-MB-453(BCRC,目錄號60429)在補充有10%FBS(HyClone,目錄號SH30071.03)、100U/mL青霉素/100μg/mL鏈霉素(GIBCO,目錄號15140)的Leibovitz's L-15培養基(GIBCO,目錄號11415)中培養。人乳腺癌細胞JIMT-1(DSMZ,目錄號ACC 589)在補充有10%FBS(HyClone,目錄號SH30071.03)、100U/mL青霉素/100μg/mL鏈霉素(GIBCO,目錄號15140)的Dulbecco's MEM培養基(GIBCO,Cat.No.11965)中培養。人胃癌細胞NCI-N87(CCRC,目錄號60217)和人T白血病細胞Jurkat(BCRC,Cat.No60424)在補充有10%FBS(HyClone,目錄號SH30071.03)、100U/mL青霉素/100μg/mL鏈霉素(GIBCO,目錄號15140)的RPMI培養基1640(GIBCO,目錄號22400)中培養。
試劑
DTT和DTPA獲自SIGMA-Aldrich(St.Louis,MO)。TCEP獲自Acros(Morris Plains,NJ)。DTNB獲自Thermo Scientific(Rockford,IL)。磷酸鈉、硼酸鈉和氯化鈉獲自J.T.Baker(Center Valley,PA)。半胱氨酸獲自Alfa Aesar(Ward Hill,MA)。
抗HER2-半胱氨酸變體的產生
將半胱氨酸殘基引入人源化抗HER2抗體(輕鏈如SEQ ID NO.9且重鏈如SEQ ID NO.10(對于IgG1)或SEQ ID NO.11(對于IgG4)),使用定向誘變方法。簡言之,通過重疊PCR進行誘變。可通過摻入核苷酸改變的引物來引入所需堿基中的具體替換。當引物延伸時,在所得擴增子中產生突變。突變位置和相應的側翼序列列于下表3中。
表3:半胱氨酸取代的突變體
表達抗HER2-Cys變體的穩定細胞系的產生
在Flp-In CHO細胞(Invitrogen,目錄號R708-07)中穩定表達和產生抗HER2半胱氨酸(抗HER2-Cys)變體(表1)。將半胱氨酸取代的抗體變體的DNA序列插入pcDNA5/FRT載體(Invitrogen,目錄號V6010-20)中,并按照供應商提供的標準程序與pOG44(Invitrogen,Cat.No V6005-20)共轉染。收集建立的細胞系的培養物上清液,并用蛋白A瓊脂糖珠子(GE Healthcare,目錄號17-5280-04)純化。純化的蛋白質用SDS-PAGE和尺寸排阻色譜法分析以確保抗體的質量。
抗HER2-IgG1抗體的常規綴合
在37℃將抗HER2-IgG1(輕鏈如SEQ ID NO.9,重鏈如SEQ ID NO.10)抗體用約0.025M硼酸鈉pH 8、0.025M NaCl、1mM DTPA中的1.55當量的TCEP還原2小時。對于1.0mg/mL溶液,使用在280nm處的吸光度值為1.48來定量蛋白質濃度,并且使用分子量145,532g/mol測定摩爾濃度。通過用DTNB滴定來測定產生的mAb-半胱氨酸硫醇的濃度。通常得到3.0硫醇/mAb。將部分還原的抗體用1.2摩爾的馬來酰亞胺己酰基藥物/mAb半胱氨酸硫醇或馬來酰亞胺-藥物(Tap18Hr1,Tap-18Hr1)/mAb-半胱氨酸硫醇烷基化。烷基化反應在4℃下進行12~16小時。半胱氨酸(1mM終濃度)用于淬滅任何未反應的過量的馬來酰亞胺基己酰基-藥物或馬來酰亞胺-藥物。Tap18Hr1綴合混合物首先用結合緩沖液、10mM磷酸鈉、10mM NaCl、5%DMSO pH7.0稀釋5倍,并以每20mg綴合的抗體1mL羥基磷灰石(稱為抗HER2/Tap18Hr1)的負載能力施加到羥基磷灰石柱(Macroprep陶瓷I型40μm,BioRad,Hercules,CA)。該柱預先用5倍柱體積的結合緩沖液平衡。施加樣品后,用3倍柱體積的結合緩沖液洗滌柱,然后用5倍柱體積的10mM磷酸鈉、10mM NaCl,pH7.0平衡。然后用200mM磷酸鈉、10mM NaCl,pH 7.0洗脫結合ADC。洗脫后,使用HiPrepTM 26/10脫鹽柱(任選)將緩沖液更換為Dulbecco's磷酸鹽緩沖鹽水。
抗HER2-Cys變體的位點特異的綴合
為了在所引入的半胱氨酸上特異性綴合連接基有效負載物(payload)Tap18Hr1(Tap-18Hr1),使用還原/氧化步驟。為了除去在培養條件下可能發生的引入的半胱氨酸位點上的半胱氨酸或谷胱甘肽,首先在37℃下在含有1mM DTPA(Sigma-Aldrich,目錄號D6518)的PBS(目錄號:21600-069)中用10~15倍摩爾過量的TCEP(Acros Organics,目錄號363830100)處理抗HER2-Cys變體2~5小時。在除去過量的TCEP后,然后將抗體用相對抗體20-70倍摩爾過量的脫氫抗壞血酸(DHA)(Sigma-Aldrich,Cat.No:261556)在室溫下再次氧化3~5小時或在4℃再次氧化3~16小時,以確保鏈間二硫鍵的重新形成。將樣品的緩沖液交換成PBS。然后加入馬來酰亞胺連接的藥物有效負載物(Tap18Hr1)以與加工的抗體上的游離巰基反應。用N-乙酰基-L-半胱氨酸(Sigma-Aldrich,Cat.No:A7250)淬滅過量的有效負載物,并使用CHT陶瓷羥基磷灰石(Bio-Rad,目錄號157-0040)純化綴合的抗體。
通過反相HPLC分析確定藥物抗體比例(DAR)
HPLC分析之前,綴合物樣品用6M胍鹽酸鹽和20mM DTT在50℃加熱下處理15分鐘。將100μg處理的綴合物樣品施加至PLRP-S柱(2.1x 150mm,8μm,Agilent)。流速為0.8mL/min且柱溫度為80℃。溶劑A為mini Q水中的0.05%三氟乙酸且溶劑B為乙腈中的0.04%三氟乙酸。該方法由以下組成:等度25%B 3ml,25ml線性梯度至50%B,2ml線性梯度至95%B,1ml線性梯度至25%B,和等度25%B 2ml。峰歸屬使用未綴合的抗體(L0和H0)進行。H1和H2通過其洗脫時間和UV譜分配(A248/280比例隨著藥物負載增加)。
抗HER2抗體和Tap18Hr1綴合物與癌細胞的結合
將1×105細胞接種在v-底96孔板中的每個孔中,并與100μl所示濃度的未綴合的Abs或ADC孵育。在4℃下孵育60~90分鐘后,將細胞用200μl FACS緩沖液(1×含有1%FBS的PBS)洗滌,用在FACS緩沖液中的100μl的1μg/ml山羊F(ab')2-抗人IgG(H+L)-RPE(Southern Biotech,目錄號2043-09)染色,然后在4℃孵育30~60分鐘。用FACS緩沖液洗滌細胞一次,并通過流式細胞術(BD LSR,BD Life Sciences)進行分析。
體外細胞毒性測試:WST-1測試
在96孔微量滴定板上,將SKOV-3細胞以每孔5x103個細胞接種,將MDA-MB-453和JIMT-1以每孔2x104個細胞接種,將NCI-N87以每孔4x104個細胞接種,并將Jurkat細胞以每孔2.5x104個細胞接種。以最終體積200μL/孔以所示濃度一式六份添加抗HER2/Tap18Hr1或未綴合抗體。然后將MDA-MB-453細胞在37℃和0%CO2下孵育96小時,在48小時更新等量培養基。SKOV-3、JIMT-1、NCI-N87和Jurkat細胞在37℃和5%CO2下孵育68-72小時。孵育后,根據制造商的說明書,通過細胞增殖試劑WST-1(Roche,目錄號11644807001)檢測細胞活力。簡言之,在孵育結束時,取出100μL培養基,并加入10μL/孔的WST-1。在最佳顯色后(當未處理對照的OD450≥1時,通過分光光度計(Molecular Devices(Sunnyvale,CA),VERSAmax酶標儀)測量450nm處的吸光度(OD450值)。獲得一式兩份的平均值,且減去背景(介質對照)。然后根據下式使用所得OD450值計算%抑制:[OD450溶劑-OD450樣品]/[OD450溶劑]*100。
癌癥異種移植模型中的ADC處理
用抗HER2/Tap18Hr1處理的SKOV-3
為了建立皮下異種移植模型,將含有25%高濃度基質膠(BD Biosciences,目錄號354248)的100μL PBS中的1x107SKOV-3細胞植入6周齡雌性C.B-17SCID小鼠的右脅腹(Lasco,Taipei,Taiwan)。在腫瘤細胞接種后大約2小時(標記為第1天)以100μL 3mg/kg靜脈內注射抗HER2/Tap18Hr1。每周用測徑器在兩個垂直維度上測量腫瘤體積一次或兩次,并根據公式(0.52*長度*寬度*寬度)計算。
用抗HER2/Tap18Hr1處理的MDA-MB-453
為了建立皮下異種移植模型,將含有50%高濃度基質膠(BD Biosciences,目錄號354248)的150μL PBS中的1x107MDA-MB-453細胞植入7周齡雌性C.B-17SCID小鼠的右脅腹(Lasco,Taipei,Taiwan)。當平均腫瘤體積達到150mm3,以100μL 3mg/kg靜脈內注射抗HER2/Tap18Hr1一次(標記為第1天)。每周用測徑器在兩個垂直維度上測量腫瘤體積兩次,并根據公式(0.52*長度*寬度*寬度)計算。
用抗HER2/Tap18Hr1和位點特異的Tap18Hr1-綴合的抗HER2半胱氨酸變體處理的NCI-N87
為了建立皮下異種移植模型,將在100μL PBS中的5x106NCI-N87細胞植入7周齡雌性C.B-17SCID小鼠的右脅腹(Lasco,Taipei,Taiwan)。當平均腫瘤體積達到180mm3,將藥物綴合的抗體靜脈內注射(標記為第1天)。每周用測徑器在兩個垂直維度上測量腫瘤體積一次或兩次,并根據公式(0.52*長度*寬度*寬度)計算。
用抗HER2/Tap18Hr1和位點特異的Tap18Hr1-綴合的抗HER2半胱氨酸變體處理的JIMT-1
為了建立皮下異種移植模型,將100μL PBS中的5x106JIMT-1細胞植入6周齡雌性C.B-17SCID小鼠的右脅腹(Lasco,Taipei,Taiwan)。當平均腫瘤體積達到100mm3,以100μL 3mg/kg靜脈內注射藥物綴合的抗體一次(標記為第1天)。每周用測徑器在兩個垂直維度上測量腫瘤體積兩次,并根據公式(0.52*長度*寬度*寬度)計算。
實施例2:基于抗HER2抗體的抗體藥物綴合物(ADC)對癌細胞的體外細胞結合活性
抗HER2/Tap18Hr1結合能力
在SKOV-3、NCI-N87、MDA-MB-453和Jurkat細胞中評價抗HER2裸抗體和Tap18Hr1-綴合的抗體的結合能力。表4中的數據顯示測試樣品顯著結合人HER2-表達細胞系(NCI-N87、SKOV-3和MDA-MB-453),但不結合不表達人HER2的Jurkat。此外,裸抗體和藥物-綴合的抗體都以相當的平均熒光強度(MFI)結合這些細胞。這些結果表明抗HER2/Tap18Hr1保持裸抗體的抗原反應性并有效地結合表達HER2的細胞。
表4:抗HER2-IgG1與癌細胞的結合
在乳腺JIMT-1、胃NCI-N87和卵巢SKOV-3癌細胞中Tap18Hr1-綴合的抗HER2半胱氨酸變體的結合能力
在乳腺JIMT-1(表5-7),胃NCI-N87(表8)和卵巢SKOV-3(表9)癌細胞中評價具有或不具有藥物綴合的抗-HER2-IgG1Cys變體的結合能力。表5-9中的數據顯示抗HER2-IgG1半胱氨酸變體與抗HER2-IgG1Ab相比同等地結合所有測試的癌細胞。此外,位點特異性綴合的抗HER2-IgG1ADC還保留了抗原反應性,除了顯示比其它變體略低的親和力的抗HER2-S442C-IgG1/Tap18Hr1。
表5.抗HER2-IgG1變體與JIMT-1細胞的結合
*ND:未測定。
表6.抗HER2-IgG1變體與JIMT-1細胞的結合
*ND:未測定。
表7.抗HER2-IgG1變體與乳腺JIMT-1癌細胞的結合
*ND:未測定。
表8.抗HER2-IgG1變體與胃NCI-N87癌細胞的結合
*ND:未測定。
表9.抗HER2-IgG1變體與卵巢SKOV-3癌細胞的結合
*ND:未測定。
在乳腺JIMT-1(表10-11)、胃NCI-N87(表12)和卵巢SKOV-3(表13)癌細胞中評價具有或不具有藥物綴合的抗HER2-IgG4p Cys變體的結合能力。表10-13中的數據顯示抗HER2-IgG4p半胱氨酸變體的結合與抗HER2-IgG4p抗體的結合相當,但與抗HER2-IgG1抗體的結合相比較低(表5-9),表明降低的結合活性歸因于IgG4同種型而不是半胱氨酸突變。總的來說,位點特異性綴合的抗HER2-IgG4p ADC保留了抗原反應性。與IgG1變體相似,抗HER2-S442C-IgG4p也顯示出比其它變體稍低的親和力。
表10.抗HER2-IgG4p半胱氨酸變體與JIMT-1細胞的結合
*ND:未測定。
表11.抗HER2-IgG1、抗HER2-IgG4p和抗HER2-IgG4p半胱氨酸變體與乳腺JIMT-1癌細胞的結合
*ND:未測定。
表12.抗HER2-IgG1、抗HER2-IgG4p和抗HER2-IgG4p半胱氨酸變體與胃NCI-N87癌細胞的結合
*ND:未測定。
表13.抗HER2-IgG1、抗HER2-IgG4p和抗HER2-IgG4p半胱氨酸變體與卵巢SKOV-3癌細胞的結合
*ND:未測定。
實施例3:基于抗HER2抗體的抗體藥物綴合物(ADC)對癌細胞的體外細胞毒性作用
在HER2陽性癌細胞系(NCI-N87,SKOV-3,MDA-MB-453和JIMT-1)和HER2陰性細胞系(Jurkat)中評價常規綴合的抗HER2抗體(抗HER2/Tap18Hr1)的體外細胞毒活性。也平行測試了裸抗體的細胞毒性。在5和1.25μg/mL,盡管抗HER2裸抗體可以誘導NCI-N87(胃癌細胞)和MBA-MD-453(乳腺癌細胞)中的細胞毒性,但常規綴合的抗HER2甚至更有效(表14)。對于JIMT-1(乳腺癌細胞)和SKOV-3(卵巢癌細胞),只有藥物綴合的抗HER2可以在5和1.25μg/mL引起超過50%的生長抑制(表14和15)。在HER2陰性細胞系Jurkat中沒有觀察到毒性。這些結果表明Tap18Hr1綴合的抗HER2遞送細胞毒性藥物到具有抗原特異性的靶癌細胞。
表14.體外細胞毒性活性
陰性值表明沒有檢測到細胞毒性。
表15.體外細胞毒性活性
還在NCI-N87、JIMT-1和SKOV-3細胞中評價了位點特異性綴合的抗HER2-Cys變體的體外細胞毒性活性。表16和17顯示了抗-HER2半胱氨酸變體的測試ADC的細胞毒性測定結果。在5和1.25μg/mL,位點特異性ADC有效殺死HER2陽性癌細胞(NCI-N87,JIMT-1和SKOV-3),但不在HER2陰性細胞系(Jurkat)中殺死細胞。盡管是稍低的結合,但抗HER2-S442C/Tap18Hr1在抗原表達細胞中誘導與其他半胱氨酸變體相似的細胞毒性程度。這些結果表明,位點特異性抗HER2ADC可以遞送細胞毒性藥物至具有抗原特異性的靶癌細胞。
表16.Tap18Hr1綴合的抗HER2-Cys變體的體外細胞毒性活性
表17.Tap18Hr1綴合的抗HER2-Cys變體的體外細胞毒性活性
實施例4:用常規綴合的抗HER2處理的SKOV-3異種移植物
在體內評價抗HER2/Tap18Hr1對卵巢癌細胞SKOV-3的功效。在腫瘤細胞接種后大約兩小時(標記為第1天),用媒介物(PBS,100μL)或100μL的300mg/kg單劑量ADC以靜脈內處理小鼠。由于包括基質膠(Matrigel)的接種體積,第1天的腫瘤大小記錄為100mm3。注射的基質膠到第15天被吸收,同時腫瘤建立并在媒介物組中穩定生長(圖4)。在第17天用抗HER2/Tap18Hr1處理抑制腫瘤生長,并且該組中的所有小鼠(5/5)顯示自第27天以來不可檢測的腫瘤。由于兩組的體重穩定地增加,觀察不到毒性。實驗表明,使用單一注射,抗HER2/Tap18Hr1可有效抑制SCID小鼠中移植的抗原陽性腫瘤的生長。
實施例5:用常規綴合的抗HER2處理的MDA-MB-453異種移植物
在體內評價抗HER2/Tap18Hr1對乳腺癌細胞MDA-MB-453的功效。當平均接種的腫瘤大小達到~150mm3時,用PBS(媒介物,100μL)或100μL的3mg/kg單劑量ADC以靜脈內處理小鼠。抗HER2/Tap18Hr1組在第8天顯示腫瘤消退,從第11天起,平均腫瘤大小進一步降低至<50mm3(圖5)。在研究結束時,6只小鼠中的3只顯示完全腫瘤消退。兩組小鼠體重均穩定增加。數據表明,抗HER2/Tap18Hr1可以有效抑制SCID小鼠中移植的抗原陽性腫瘤的生長。
實施例6:用Tap18Hr1常規綴合的抗HER2和位點特異的綴合的抗HER2-Cys變體處理的NCI-N87異種移植物
在體內對胃癌細胞NCI-N87評價常規綴合的抗HER2/Tap18Hr1的功效。當平均腫瘤大小達到~180mm3時,用PBS(媒介物,100μL)或ADC(3或5mg/kg,100μL)靜脈內處理小鼠兩次(標記為第1天和第22天)。盡管媒介物組的腫瘤生長并在第15天接近500mm3(圖6),但抗HER2/Tap18Hr1組在第5天顯示延遲的腫瘤生長,從第19天開始,平均腫瘤大小進一步降低至<30mm3。在研究結束時,大多數腫瘤在兩個ADC治療組中低于10mm3。小鼠體重在三組中穩定增加。數據表明,抗HER2/Tap18Hr1可以有效抑制SCID小鼠中移植的抗原陽性腫瘤的生長。
在體內對胃癌細胞NCI-N87評價了位點特異性綴合的抗HER2-Cys變體的功效。當腫瘤大小的平均值達到~180mm3時,用PBS(媒介物,100μL)或具有相等藥物劑量(9.7μg/kg Tap18Hr1在100μL中)的ADC靜脈內處理小鼠(標記為第1天)。如圖7所示,與媒介物組相比,所有位點特異性綴合的變體處理的小鼠顯示顯著延遲的腫瘤生長。體重在ADC治療組中保持不變,并且由于腫瘤的重量,在媒介物組中輕微增加。數據證明,通過單次注射,位點特異性綴合的抗HER2-Cys變體可有效抑制在SCID小鼠中移植的抗原陽性腫瘤的生長。
實施例7:用Tap18Hr1常規綴合的抗HER2和位點特異的綴合的抗HER2-Cys變體處理的JIMT-1異種移植物
在體內針對已知的赫賽汀抗性乳腺癌細胞JIMT-1評價常規綴合的抗HER2/Tap18Hr1和位點特異性綴合的抗HER2-Cys變體的功效。當平均腫瘤大小達到~100mm3時,用PBS(媒介物對照,100μL)或ADC(3mg/kg,在100μL中)靜脈內處理小鼠一次(標記為第1天)。如圖8所示,與媒介物組相比,所有ADC治療組顯示自第7天以來顯著延遲的腫瘤生長。小鼠的體重在所有組中穩定增加。數據表明,單次給藥,不僅常規抗HER2/Tap18Hr1而且位點特異性綴合的抗HER2-Cys變體都可以有效抑制在SCID小鼠中移植的HER2陽性腫瘤的生長。
- 還沒有人留言評論。精彩留言會獲得點贊!