醇鎂法制備4-甲基兒茶酚的制作方法
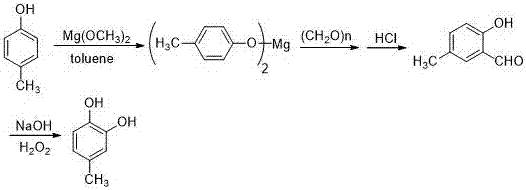
本發明采用對甲苯酚為原料,經與金屬甲醇鎂反應制備對甲苯酚鎂,對甲苯酚鎂與多聚甲醛反應制備3-甲基水楊醛,用雙氧水氧化3-甲基水楊醛制得4-甲基兒茶酚,以對甲酚計,反應總收率可達80%。與現有文獻相比,該法具有工藝路線簡單、反應條件溫和、反應收率高和適宜工業化等優點。
背景技術:
4-甲基兒茶酚是一重要的基礎化工原料,其合成方法有很多文獻報道,綜述文獻,其合成方法大致有如下幾類:1.液化木質素法。該法采用高溫(370-380oc)和高壓(200-210atm),用三氧化二鐵、氧化鎳和硫(三者的物質量比為fe:ni:s=20:1:21)為催化劑反應1hr,反應收率只有11%[6]。2.對甲酚直接氧化法。直接氧化法合成4-甲基兒茶酚的文獻報道較多[9-11,13-16],直接氧化法采用高濃度的雙氧水(大于60%),用無機酸和金屬絡合物等做催化劑。該類方法一步合成4-甲基兒茶酚,但采用高濃度雙氧水,工業生產很不安全;此外該類方法普遍存在收率不高,未反應的對甲酚及副產物很難分離。3.酶催化劑生物化學法[24-26]。該類方法是所有合成方法中最環保的,但酶催化劑的成本高,難以工業化生產。4.fries重排法[5,36,37]。以對甲基苯酚為原料,經醋酐酯化、fries重排和過氧化氫氧化合成對甲基兒茶酚。該方法fries重排步驟采用無水三氯化鋁為催化劑,反應所需溶劑量很大;此外后處理三氯化鋁產生大量對設備腐蝕極大的氯化氫氣體和大量廢水。5.酚鎂-甲醛法[38-40]。該方法用無水氯化鎂為催化劑,三乙胺為縛酸劑,經與多聚甲醛反應合成3-甲基水楊醛,然后用雙氧水氧化3-甲基水楊醛合成4-甲基兒茶酚。該方法收率、反應步驟與fries重排法類似,由于無水氯化鎂水解比無水三氯化鋁溫和,無氯化氫氣體產生,但也存在反應所需溶劑量很大,產生大量的廢水等缺點,此外三乙胺的回收增加了生產成本和廢水的產生。受酚鎂-甲醛法啟發,我們在合成3-甲基水楊醛時,用對甲酚與金屬鎂反應合成對甲酚鎂。與酚鎂-甲醛相比,該方法無需縛酸劑,固液比小,所需溶劑量小。具有工藝路線簡單、反應條件溫和、反應收率高和適宜工業化等優點。
技術實現要素:
本發明要解決的技術問題是4-甲基兒茶酚的合成。以對甲酚為原料,經與金屬鎂反應合成對甲酚鎂,以對甲酚鎂合成3-甲基水楊醛,然后經雙氧水氧化合成4-甲基兒茶酚。說明書附圖中的圖1是4-甲基兒茶酚的合成路線圖。
實例一
將鎂粉(14.4g,0.6mol)投入裝有200ml甲醇的四口反應瓶中,開啟電動攪拌,瓶內產生大量氣泡,溫度上升至61℃。2h后,溶液變成乳白色懸濁液。將對甲基苯酚(108g,1.0mol)加到反應瓶中,50min后開始蒸出甲醇,待反應液變稠,補200ml甲苯。升高溫度,待內溫達到95℃時,分批加入多聚甲醛(90g,3.0mol),將裝置改為蒸餾裝置,蒸出低沸點物質,30min后幾乎無餾分蒸出,撤去蒸餾裝置,繼續反應2.5h,用tlc點板跟蹤,對甲基苯酚反應完全。停止加熱,待內溫降至65℃,用18.5%的鹽酸調節ph=1。靜置后分液,有機層用70ml甲苯萃取2次,合并有機層,有機層水洗3次至ph接近7。得到中間體4-甲基水楊醛。將中間體4-甲基水楊醛投入裝有80ml水,250ml工業酒精和110ml雙氧水的反應瓶中,開啟電動攪拌,在冰浴下緩慢滴加30%氫氧化鈉,并維持溫度在18℃左右。用tlc點板跟蹤,2h后反應完全。滴加20%亞硫酸鈉淬滅雙氧水,使碘化鉀淀粉試紙不變藍時,用18.5%的鹽酸調節ph至2-3之間。然后100ml乙酸乙酯萃取4次,合并有機層。旋干溶劑得到4-甲基兒茶酚粗品。用乙酸乙酯(v):石油醚(v)=7:3為重結晶溶劑,得到4-甲基兒茶酚白色固體87克,熔點64-66℃,純度99.78%(tc,外標)收率(以對甲酚計)69.9%。1hnmr和13cnmr(500mhz,cdcl3)如下:1hnmr(cdcl3,500mhz)δ6.75(d,j=8.0hz,1h),6.70(d,j=1.0hz,1h),6.61(s,j=8.0hz,1h),5.24(s,2h),2.60(s,3h);13cnmr(cdcl3,125mhz)δ143.3,141.0,131.0,121.4,116.2,115.3,20.7。
技術特征:
技術總結
4?甲基兒茶酚是一重要的基礎化工原料,其合成方法有很多文獻報道,但這些方法難以工業化。本發明以對甲酚為原料,經與醇鎂反應合成對甲酚鎂,以對甲酚鎂合成3?甲基水楊醛,然后經雙氧水氧化合成4?甲基兒茶酚。以對甲酚計,反應總收率可達80%。與現有文獻相比,該法具有工藝路線簡單、反應條件溫和、反應收率高和適宜工業化等優點。
技術研發人員:劉建兵;祝岱楨;黃雅丹
受保護的技術使用者:湖南師范大學
技術研發日:2016.03.21
技術公布日:2017.09.29
- 還沒有人留言評論。精彩留言會獲得點贊!